The Annotated AlphaFold2
A line-by-line walkthrough of Jumper et al. 2021, with a minimal PyTorch implementation you can actually run.
AI x biology should be one of the most important technical fields of the next decade: the path to better medicines, better diagnostics, better protein and enzyme design, and a much deeper ability to intervene on disease. And yet the path into the field is still strangely illegible. Compared with LLMs, coding agents, or image generation models, biological AI has very few clear walkthroughs, minimal implementations, or pedagogical codebases.
This matters because talent flows toward legible fields. When the path into biological AI is harder to see, fewer engineers and researchers enter it, which means fewer projects, fewer companies, less venture funding, and slower downstream medical innovation. This site is my attempt to make that path more legible by pairing important biological AI papers with runnable, paper-aligned implementations.
The right place to start is AlphaFold2. It was the first major deep learning breakthrough in biology and, in my view, probably the second most important deep learning model in history after GPT-3. It changed structural biology, won the Nobel Prize, and became the root node for much of what followed. Once you understand AlphaFold2, later papers become much easier to place: some modify its MSA machinery, some replace it with protein language models, some extend the structure module, and some broaden the task from structure prediction to sequence-structure-function modeling.
So I wrote minAlphaFold2: a pedagogical PyTorch reimplementation designed to be read alongside the 62-page AlphaFold2 supplement. Variable names match the paper’s notation, comments cite the supplement’s algorithm numbers inline, and every major component below is tied back to runnable code. The goal is not production deployment. The goal is understanding.
If you’d like to follow along locally, clone the repo and run
pip install -e .. Thescripts/overfit_single_pdb.pytrainer runs on a laptop CPU and takes a small protein from random initialization to roughly 2 Å Cα RMSD in about thirty seconds. We’ll walk through that exact run in §14. Let’s dive in.
0. Prelims
Throughout this guide, we’ll make use of minAlphaFold2, which is a simple, easy-to-read re-implementation of AlphaFold2 that I produced. You can access the repo here: https://github.com/ChrisHayduk/minAlphaFold2
The goal of minAlphaFold2 is to make AlphaFold2 simple and legible. The full DeepMind implementation is a production-oriented JAX codebase with tons of bells and whistles that make it extremely confusing to dig through. On the other hand, minAlphaFold2 is a much smaller pure-PyTorch implementation written specifically to be read against the paper. Every design decision in the repo follows from that North Star:
Variable names follow the paper exactly. m is the MSA representation, with shape , where is the number of sequences, is the number of residues, and is the channel dimension. z is the pair representation, with shape . s_i is the single representation in the Structure Module. T_i is a rigid frame. If you have the AF2 paper and supplement open, you should be able to jump back and forth between the papers and the code with no cognitive tax,
Algorithms are annotated inline. Every major class and function in minAlphaFold2 cites the supplement algorithm it implements. Comments like # Algorithm 7 appear right above the relevant forward method, allowing you to reference the description and pseudocode from the paper while reading the code. When I say “Algorithm 7” below, you can grep for it in the repo to find the corresponding code. Shapes are typically asserted throughout the forward pass, and the tests exercise those assertions end-to-end.
There are three config profiles living under configs/, intended to provide runnable versions of minAlphaFold2 for a range of compute budgets:
tiny.toml— about 90k parameters, CPU-runnable, good for unit tests and for the overfit walkthrough in §14.medium.toml— for overfit experiments on a single GPU.alphafold2.toml— the full 48-block, paper-spec config.
The walkthrough below will mostly stay at the level of the architecture, but when we get to the concrete example in §14, we’ll load tiny.toml and watch the Structure Module actually converge on a real protein.
1. Background
AlphaFold2 was not the first deep-learning approach to protein structure prediction, but it was groundbreaking because it was the first deep learning system to really work. It produced structures that were far more accurate than any other existing computational methods, approaching the accuracy levels of experimental structure determination for a large fraction of proteins. To understand why it worked, we need to start with the biology rather than with the architecture, because the architecture only makes sense once you understand the particular statistical structure the model is exploiting.
1.1 What are proteins
Let’s start with a seemingly simple question - what are proteins and why should we care about them?
Proteins are large molecules produced within the cells of living organisms to perform some function. You can think of proteins as the machinery of life. They are responsible for facilitating chemical reactions in the body, providing structural integrity to cells, acting as receptors, and more. Quite simply, life as we know it would not be possible without the vast array of protein-based nanomachines constantly at work in our bodies.
The functions that a protein takes on (e.g., whether it acts as a receptor attached to a cell membrane or as a catalyst for a chemical reaction) come down in large part to its 3D structure. The shape of the protein determines what other molecules it can interact with, thereby determining what it can do in the body.
Now, if proteins are so important, it may be natural to ask “where do they come from?” And herein lies the central role of DNA in living organisms.
It is a common cliché to hear that DNA is the system of record for an organism, or the set of instructions for life. But it is less often said exactly what instructions DNA is providing.
As it turns out, DNA is essentially a set of instructions for making proteins! The role of DNA is to tell your cell what proteins to make, when to make them, and in what quantities to make them. In fact, there is a clear mapping between DNA and proteins - if you remember your biology, you’ll remember that DNA is made up of base pairs (represented by the letters ACTG in a long string). Chunks of three of these base pairs, referred to as “codons”, are translated by the ribosomes in your cells into amino acids. So much of what your body does is converting strings of DNA base pair sequences into strings of amino acid sequences.
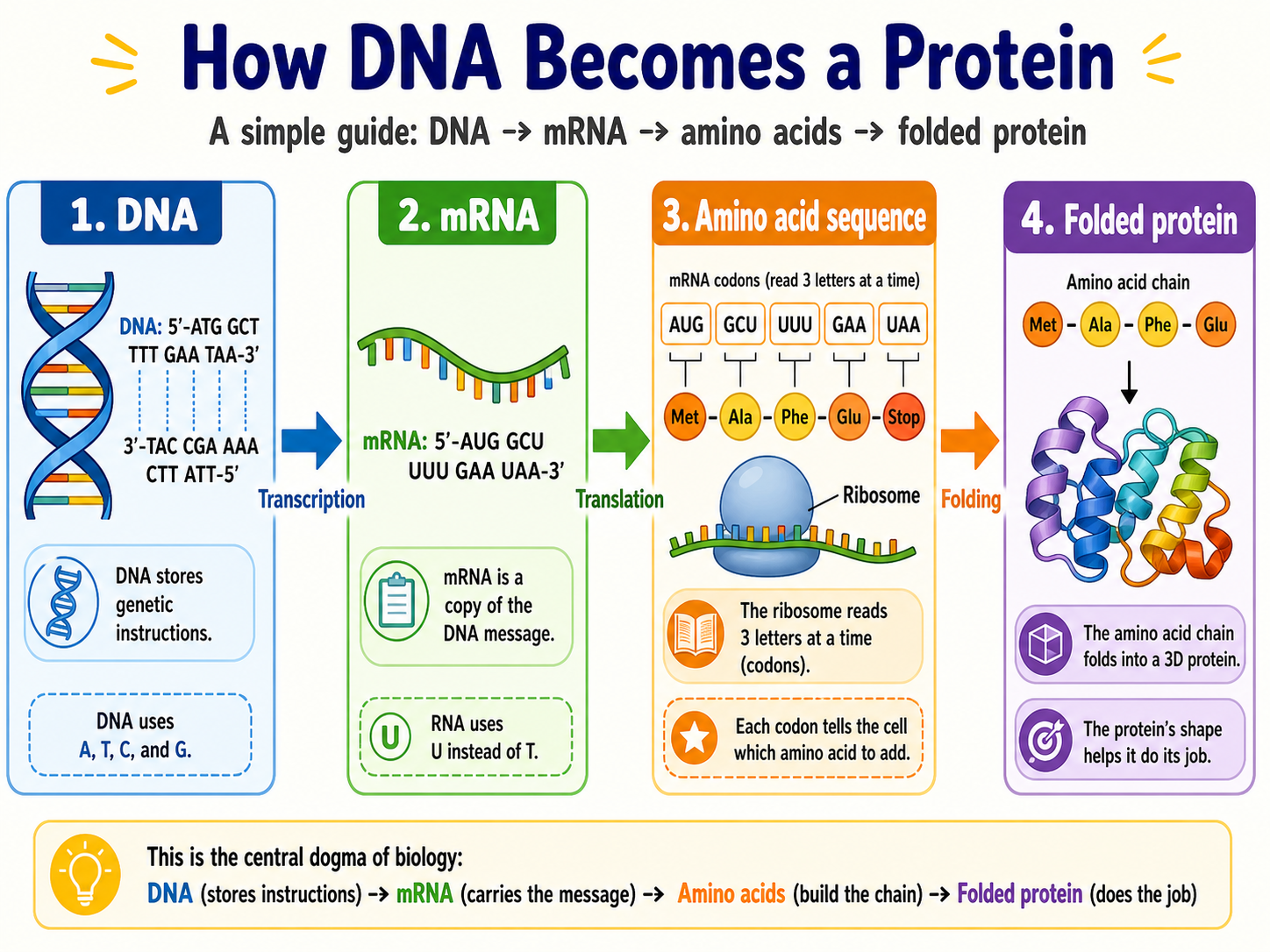
Hence, from two different views, a protein is simultaneously a one-dimensional string of amino acids and also a three-dimensional, interactive structure. How can we reconcile these two views?
They’re connected by what’s known as protein folding.
1.2 The protein folding problem
Protein folding is the process by which the one-dimensional amino acid sequence output by the ribosomes folds into its ultimate 3D shape. As we discussed earlier, this shape determines the functions that the protein can fulfill in the body. Thus, function follows structure, and structure follows sequence.
In practice, determining a protein’s amino acid sequence is much easier, faster, and cheaper than determining its 3D structure, so it would be nice to be able to predict the structure from the sequence. Unfortunately, this task was historically nearly impossible.
The protein folding problem is hard for the reason Cyrus Levinthal pointed out in 1969 - a sequence of length 100 has something like possible conformations if each residue has three rotational states. The search space is so large that it is impossible to brute force, regardless of any realistically conceivable amount of compute thrown at it. And yet proteins fold on millisecond timescales inside cells, reliably and reproducibly!
The only way this can happen is if the energy landscape is highly biased. Rather than sample uniformly from all possible conformations, proteins are being funneled by the physics of their interactions toward a small set of low-energy structures.
This insight opens the door to modeling the funneling dynamics present in protein folding. For decades, the field tried to model that funnel directly through molecular dynamics, statistical potentials, fragment libraries, and many other approximations. These methods worked in pieces, but not well enough at the proteome scale due to their immense computational cost. Running a full molecular dynamics simulation for large proteins was just computationally intractable.
AlphaFold2’s core innovation was to stop trying to explicitly simulate the entire physical process and instead learn the shape of the funnel from data. We’ll discuss two of these key data sources in the next two subsections (1.2 and 1.3).
1.3 MSAs and evolutionary coupling
The key insight that unlocks protein folding, which predates AlphaFold2 by decades, is that related proteins across species tend to share the same domains, and the mutations they accumulate over evolutionary time are not independent. If amino acids and in the protein sequence are in contact in the 3D structure, then a mutation at that disrupts that contact creates selection pressure for a compensating mutation at . That is, if doesn’t mutate to compensate for the mutation at , then the protein won’t successfully fold into the shape needed to achieve its function. Over millions of years and many species, that pressure leaves a statistical signature - the columns for residues and in a multiple sequence alignment co-vary.
A multiple sequence alignment, or MSA, is exactly what it sounds like. We take the target sequence, find a large set of evolutionarily related sequences, and align them so that homologous residues are in the same column.
(Terminology note: homologous == “evolutionarily related positions in the amino acid sequences”, residue == “a single amino acid in a sequence”)
We can see an example of a small MSA below. The rows represent related proteins from various species. Each column is an aligned position in those sequences (e.g., amino acid 1 is in the first column).
H. sapiens - - - - - G D V E K G K K I F I M K C S Q C H E. caballus - - - - - G D V E K G K K I F V Q K C A Q C H G. gallus - - - - - G D I E K G K K I F V Q K C S Q C H D. melanogaster - G V P A G D V E K G K K L F V Q R C A Q C H S. cerevisiae T E F K A G S A K K G A T L F K T R C L Q C H
From an MSA, you can infer a surprising amount. Column variation tells you which residues are conserved, meaning evolution has decided they matter, and which residues are free to drift.
(Another terminology note: in biology speak, “conserved residues” just means amino acids in the same position that don’t change across species in the MSA.)
Correlated columns tell you which residues are coupled, which is often the signature of contact in 3D. This is why AlphaFold2 treats the MSA as a first-class input rather than as a side feature - these coupled residues give the model a strong starting point to “guess” which residues should be next to each other in the 3D structure.
The claim that co-evolution equals contact deserves to be seen rather than asserted because it forms part of the core for why AlphaFold works and is critical to understanding its architectural choices. The widget below lays out an MSA of about 2,000 serine proteases (Pfam PF00089, aligned with MAFFT), the standard contact-prediction heatmap (top-L coupled pairs in red vs. top-L real contacts in blue — the same evaluation format DCA papers use), and the real bovine β-trypsin structure (2PTN), all linked together. Click any cell, or pick a preset below, and the corresponding pair lights up in all three panels. Bright red cells are coupling predictions that landed on a true contact; faded red cells are false positives. The signal here genuinely useful! Roughly a quarter of the top-L predictions are real contacts, several multiples of the random baseline. That’s what AlphaFold2 inherits as raw material and sharpens with the Evoformer.
AlphaFold2 builds its MSAs by searching four databases with two tools (HHBlits for sensitivity, JackHMMER for coverage):
- MGnify — metagenomic sequences, where a lot of novel protein diversity lives.
- UniRef90 — clustered UniProt, the main protein sequence database.
- Uniclust30 — another clustering of UniProt, at 30% sequence identity.
- BFD — Big Fantastic Database, billions of sequences assembled from raw metagenomic reads.
For our purposes, the MSA pipeline & algorithms themselves are out of scope. Instead, minAlphaFold2 takes a pre-built MSA as input. What matters is the shape of what comes out: a tensor of one-hot-ish per-residue features for each aligned sequence, typically a few hundred to a few thousand sequences deep. Everything we do from here is downstream of that tensor.
If you’d like to go deeper on MSAs specifically (how AF2 uses row- and column-wise attention on the MSA representation, and why the outer product mean is the right way to push that information into the pair representation) I’ve written a three-part primer on protein language models that covers the MSA pipeline in Part I and the transformer-based replacement (ESMFold) in Parts II and III. This annotation will cover all of that ground too, but from the minAlphaFold2 codebase up rather than from the paper down.
1.4 Templates
With MSAs, we are looking at sequences that are evolutionarily similar to our target sequence in order to find correlations in individual amino acids. But if we already know the solved 3D structure of one of these similar proteins? If its sequence is very close to our target, shouldn’t its structure be close as well?
This is where templates come in, forming the second source of prior knowledge AlphaFold2 uses. If an evolutionarily related protein already has a solved 3D structure in the Protein Data Bank, its coordinates can be used as a hint for the model to “guess” at the coordinates of the target protein. AlphaFold2 finds up to four templates via HHSearch on PDB70, converts them into pair-like embeddings, and injects them into the same broad pipeline as the MSA (i.e., the Evoformer).
Hence, while we can think of MSA as giving us granular information on which pairs. of residues are in contact, we can think of templates as giving us global information about what the entire folded protein might look like.
In practice within AlphaFold2, templates are mostey useful when evolutionary information is sparse (that is, when the MSA search for a protein does not return many hits), but the model does not depend on them in the way older template-based methods did. The MSA and the learned pair representation do most of the work. As such, we will treat templates as an important input-pipeline detail and cover them briefly in §8, rather than making them the center of the story.
1.5 Rigid frames and SE(3)
The output of AlphaFold2 is a 3D structure, but the most important intermediate object is not just a list of Cartesian coordinates. Instead, each residue has an associated local frame: a position and a rotation that together specify a small coordinate system attached to the backbone. The whole structure can then be represented as a sequence of these frames, one per residue.
This matters for two reasons. First, frames are the natural object for expressing geometric invariances. The physics of protein folding does not care about the global position or orientation of the molecule. If you translate and rotate the entire structure, it is still the same protein. That is SE(3) invariance, and any loss or model component that respects it has a much easier problem than one that has to learn it from scratch. Second, frames give us a clean way to express local geometry. The position of residue as seen by residue is just , meaning the translation expressed in residue ‘s local coordinate system. That “as seen by” operation is the core of Invariant Point Attention, which we’ll get to in §10.
For §§1–9, we mostly won’t need to touch frames. They will show up properly when we reach the Structure Module. But it is worth keeping in mind, as we watch the Evoformer update an abstract pair representation , that the whole upstream model is preparing the information the Structure Module will need in order to place atoms in space.
1.6 What AlphaFold2 changed
Before diving into the implementation, it is useful to be explicit about what was actually new. The rough outline of a contact-prediction-plus-folding pipeline had existed for years: run an MSA, extract co-evolution signals, predict a residue-residue contact map, then fold the protein through fragment assembly or distance-based minimization. AlphaFold1 was already doing something in this family. AlphaFold2 changed the game by adding the following:
- End-to-end training. No separate “predict contacts, then fold” pipeline. The entire system, from MSA input to atom coordinates, is one computation graph with one loss.
- The Evoformer. A transformer-style stack that cross-attends between the MSA representation and the pair representation, letting each update the other over 48 blocks.
- Invariant Point Attention. A geometric attention variant that operates on rigid frames, respects SE(3) invariance by construction, and lets the Structure Module reason about 3D without losing equivariance.
- FAPE loss. A frame-aligned loss that measures error per-residue-frame rather than in a global coordinate system, giving every residue a local view of the structure it’s trying to build.
- Recycling. A mechanism where the model’s own output predictions get fed back as input for another forward pass, enabling iterative refinement at both train and test time.
Each of these gets its own section. The rest of this article is essentially a tour of how these five ideas become executable PyTorch. Before we zoom in, here is the full pipeline at a glance: three input modalities on the left, the two-stream Evoformer in the middle, the Structure Module turning representations into atoms, and a recycling loop wrapping the whole thing. Every box below points to the section where we will open it up.
2. Input representations
At this point, we need to convert biology into tensors. AlphaFold2 takes in three kinds of raw data — the target sequence, the MSA, and the templates — and distills them into two working representations that flow through the rest of the network:
- the MSA representation , shape , where is the number of sequences in the alignment and is the number of residues.
- the pair representation , shape — one embedding for every pair of residues in the target sequence.
You can think of as “what we know about each residue, as seen through evolutionary history” and as “what we know about each pair of residues.” The whole Evoformer is built around passing information back and forth between these two objects: using the pair representation to guide attention within the MSA, and using the MSA to refine the pair representation via the outer product mean. We’ll get there shortly.
2.1 Building the features
The feature builders that turn raw inputs — an integer-encoded sequence, a stack of aligned MSA sequences, and a set of template structures — into embedding-ready tensors live in build_target_feat . Most of this is one-hot encoding and concatenation. That may sound boring, but it matters because these feature tensors define what the network is allowed to know. The headliners are:
-
build_target_feattakes the target integer-encoded sequence and produces a per-residue feature vector: a one-hot over the 21 amino-acid types (20 + unknown), plus a per-residue “deletion value” and a few binary flags. Dimension 22. -
build_msa_feattakes the full MSA and produces a tensor — for each sequence and each residue, a one-hot over 23 MSA tokens (the 20 amino acids, plus gap, mask, and unknown), a cluster profile averaged across the sequences in that cluster, plus features recording which residues had gap deletions and how many. This is where evolutionary information enters the model. -
build_extra_msa_featdoes the same thing for the “extra MSA” stack — a larger, cheaper-to-process slice of the alignment that gets its own shallower Evoformer (covered in §8).
2.2 The input embedder
Once we have the feature tensors, the InputEmbedder block projects them into and (Algorithm 3 of the supplement). The math is straightforward: three linear projections of the target features plus an outer-sum to produce a per-pair embedding for ; one linear projection of the MSA features to produce ; and a tiled copy of the target features added across all MSA rows.
class InputEmbedder(torch.nn.Module):
"""Initial MSA + pair embedding (Algorithm 3).
Produces the starting ``m_si`` and ``z_ij`` for the Evoformer by
combining:
* three linear projections of ``target_feat`` (shape
``(batch, N_res, TARGET_FEAT_DIM)``) — two broadcast into the
outer-sum for ``z`` and one added to the query row of ``m``;
* a relative-positional encoding ``RelPos(residue_index)`` added to
``z`` (Algorithm 4);
* a linear projection of ``msa_feat`` (49 channels — cluster profile
+ deletion features, per Table 1) added to every MSA row in ``m``.
Output shapes: ``m`` ``(batch, N_cluster, N_res, c_m)``, ``z``
``(batch, N_res, N_res, c_z)``.
"""
def __init__(self, config):
super().__init__()
self.linear_target_feat_1 = torch.nn.Linear(in_features=TARGET_FEAT_DIM, out_features=config.c_z)
self.linear_target_feat_2 = torch.nn.Linear(in_features=TARGET_FEAT_DIM, out_features=config.c_z)
self.linear_target_feat_3 = torch.nn.Linear(in_features=TARGET_FEAT_DIM, out_features=config.c_m)
self.linear_msa = torch.nn.Linear(in_features=49, out_features=config.c_m)
init_linear(self.linear_target_feat_1, init="default")
init_linear(self.linear_target_feat_2, init="default")
init_linear(self.linear_target_feat_3, init="default")
init_linear(self.linear_msa, init="default")
self.rel_pos = RelPos(config)
def forward(self, target_feat: torch.Tensor, residue_index: torch.Tensor, msa_feat: torch.Tensor):
# target_feat shape: (batch, N_res, 22)
# residue_index shape: (batch, N_res)
# msa_feat shape: (batch, N_cluster, N_res, 49)
assert target_feat.ndim == 3 and target_feat.shape[-1] == TARGET_FEAT_DIM, \
f"target_feat must be (batch, N_res, {TARGET_FEAT_DIM}), got {target_feat.shape}"
assert residue_index.ndim == 2, \
f"residue_index must be (batch, N_res), got {residue_index.shape}"
assert msa_feat.ndim == 4 and msa_feat.shape[-1] == 49, \
f"msa_feat must be (batch, N_cluster, N_res, 49), got {msa_feat.shape}"
# Output shape: (batch, N_res, c_z)
a = self.linear_target_feat_1(target_feat)
b = self.linear_target_feat_2(target_feat)
# Output shape: (batch, N_res, N_res, c_z)
# Row i should use element i from a, and col j should use element j from b
z = a.unsqueeze(-2) + b.unsqueeze(-3)
z += self.rel_pos(residue_index)
# Output shape: (batch, N_cluster, N_res, c_m)
m = self.linear_target_feat_3(target_feat).unsqueeze(1) + self.linear_msa(msa_feat)
return m, zTwo details are worth lingering on. First, the pair representation is initialized from an outer-sum of the target features, not from the MSA. The MSA features arrive later through the outer product mean inside the Evoformer. Right after the embedder, does not yet know anything about co-evolution; it is a target-sequence scaffold that the Evoformer will fill in. Second, the target features are tiled across every row of , including rows corresponding to other MSA sequences. This is the model’s way of telling each row, “you are aligned to this target,” which gives the row attention and column attention operations a shared coordinate system.
2.3 Relative position encoding
The second piece of the input-embedding stage is the relative position encoding (Algorithm 4). Like the positional encoding in a vanilla Transformer, this tells the network where each residue sits in the chain. But AlphaFold2 encodes relative positions rather than absolute ones, which fits the problem better: what matters structurally is whether two residues are adjacent, nearby, or far apart along the chain, not their absolute index in some fixed-length array.
class RelPos(torch.nn.Module):
"""Relative-position encoding (Algorithm 4).
One-hots the clipped residue-index difference
``clamp(r_i - r_j, -max_rel, max_rel)`` into ``2·max_rel+1`` bins
(default ``max_rel = 32`` → 65 bins) and projects to ``c_z``. The
output is added to the pair representation by :class:`InputEmbedder`
so the Evoformer trunk has a learned sense of residue adjacency from
the very first block. Clipping at ±32 matches the supplement.
"""
def __init__(self, config, max_rel=32):
super().__init__()
self.max_rel = max_rel
self.linear = torch.nn.Linear(2 * max_rel + 1, config.c_z)
init_linear(self.linear, init="default")
def forward(self, residue_index: torch.Tensor):
# residue_index shape: (batch, N_res)
d = residue_index[:, :, None] - residue_index[:, None, :] # (batch, N_res, N_res)
d = d.clamp(-self.max_rel, self.max_rel) + self.max_rel
oh = torch.nn.functional.one_hot(d.long(), 2 * self.max_rel + 1).float()
return self.linear(oh) # (batch, N_res, N_res, c_z)The encoding is a clipped one-hot over within a window of residues. Anything further away gets bucketed to the edge bins. This one-hot vector is then linearly projected to and added directly into . This is simple, but important. Without it, the pair representation has no built-in notion of sequence adjacency, and the model would have to rediscover the protein chain from the other pair features alone.
3. The Evoformer — bird’s-eye view
The Evoformer is the core of AlphaFold2. It is a stack of 48 blocks, each one taking an MSA representation and a pair representation , applying a fixed sequence of updates, and passing the refined to the next block. By the end of the stack, and are dense with structure-relevant information and ready for the Structure Module to consume.
Before opening a block up, it helps to have the topology in front of us. The diagram below shows one block: two streams running left-to-right, seven sub-blocks in temporal order, and two cross-stream arrows that capture the whole architectural idea.
class Evoformer(torch.nn.Module):
"""Evoformer block (Algorithm 6).
One full iteration over the paired MSA + pair representations:
1. MSA row-wise attention with pair bias (Alg 7) + row-wise dropout;
2. MSA column-wise attention (Alg 8) — no dropout;
3. MSA transition (Alg 9) — no dropout;
4. Pair update from the MSA via outer-product mean (Alg 10);
5. Triangle multiplicative updates outgoing/incoming (Alg 11/12) +
row-wise dropout;
6. Triangle self-attention around the starting/ending node (Alg 13/14)
— row-wise dropout on starting, column-wise on ending
(supplement 1.11.6);
7. Pair transition (Alg 15) — no dropout.
Dropout rates are read from ``config.evoformer_msa_dropout`` and
``config.evoformer_pair_dropout`` so the Template pair stack
(:class:`minalphafold.embedders.TemplatePair`) and the Extra MSA stack
(:class:`minalphafold.embedders.ExtraMsaStack`) can reuse the same
sub-modules with different dropout schedules. The block is stacked
``config.num_evoformer_blocks`` times in :class:`minalphafold.model.AlphaFold2`.
"""
def __init__(self, config):
super().__init__()
self.msa_row_att = MSARowAttentionWithPairBias(config)
self.msa_col_att = MSAColumnAttention(config)
self.msa_transition = MSATransition(config)
self.outer_mean = OuterProductMean(config)
self.triangle_mult_out = TriangleMultiplicationOutgoing(config)
self.triangle_mult_in = TriangleMultiplicationIncoming(config)
self.triangle_att_start = TriangleAttentionStartingNode(config)
self.triangle_att_end = TriangleAttentionEndingNode(config)
self.pair_transition = PairTransition(config)
# Dropout rates from config
self.msa_dropout = config.evoformer_msa_dropout
self.pair_dropout = config.evoformer_pair_dropout
def forward(self, msa_representation: torch.Tensor, pair_representation: torch.Tensor,
msa_mask: Optional[torch.Tensor] = None, pair_mask: Optional[torch.Tensor] = None):
# msa_mask: (batch, N_seq, N_res) — 1 for valid, 0 for padding
# pair_mask: (batch, N_res, N_res) — 1 for valid, 0 for padding
assert msa_representation.ndim == 4, \
f"msa_representation must be (batch, N_seq, N_res, c_m), got {msa_representation.shape}"
assert pair_representation.ndim == 4, \
f"pair_representation must be (batch, N_res, N_res, c_z), got {pair_representation.shape}"
# Shape (batch, N_seq, N_res, c_m)
z = self.msa_row_att(msa_representation, pair_representation, msa_mask=msa_mask)
msa_representation = msa_representation + dropout_rowwise(z, p=self.msa_dropout, training=self.training)
# No dropout on column attention or MSA transition per Algorithm 6
msa_representation = msa_representation + self.msa_col_att(msa_representation, msa_mask=msa_mask)
msa_representation = msa_representation + self.msa_transition(msa_representation)
pair_representation = pair_representation + self.outer_mean(msa_representation, msa_mask=msa_mask)
pair_representation = pair_representation + dropout_rowwise(self.triangle_mult_out(pair_representation, pair_mask=pair_mask), p=self.pair_dropout, training=self.training)
pair_representation = pair_representation + dropout_rowwise(self.triangle_mult_in(pair_representation, pair_mask=pair_mask), p=self.pair_dropout, training=self.training)
pair_representation = pair_representation + dropout_rowwise(self.triangle_att_start(pair_representation, pair_mask=pair_mask), p=self.pair_dropout, training=self.training)
pair_representation = pair_representation + dropout_columnwise(self.triangle_att_end(pair_representation, pair_mask=pair_mask), p=self.pair_dropout, training=self.training)
# No dropout on pair transition per Algorithm 6
pair_representation = pair_representation + self.pair_transition(pair_representation)
return msa_representation, pair_representationEach block does, in order: (1) MSA row-wise attention biased by , (2) MSA column-wise attention, (3) an MSA transition MLP, (4) an outer product mean that writes MSA information back into , (5) triangle multiplicative updates (outgoing and incoming) on , (6) triangle self-attention (starting-node and ending-node) on , and (7) a pair transition MLP. That is seven sub-blocks per Evoformer block, 48 blocks total, and every sub-block has a dropout pattern matched to its shape.
It is worth noticing how lopsided this is. Of the seven sub-blocks, only the first three operate on ; the rest operate on or write into it. The MSA stack is relatively narrow and mostly one-way: MSA → pair via the outer product mean. The pair stack is where most of the geometry-like reasoning happens: triangle consistency, triangle-biased attention, and pairwise transitions. In essence, the design is: extract co-evolution signal from the MSA, push it into the pair representation, then spend most of the block refining the pair representation into something the Structure Module can use.
Two structural details are important before we dive in. First, the MSA stack terminates at the end of the Evoformer. Only the first row of , corresponding to the target sequence, is projected into the single representation and passed to the Structure Module. The other rows have done their job: they have pushed their evolutionary signal through the outer product mean and into . Second, recycling (§9) wraps around the whole model. The output of block 48 becomes part of the input of block 1 in the next cycle, and this can happen up to four times per forward pass in the paper setup. Hence, “48 blocks” is really “up to 192 blocks of compute,” but with weights shared across recycling cycles.
Now let’s open up the Evoformer block sub-block by sub-block.
4. MSA row attention with pair bias
This is Algorithm 7 of the supplement, and it is one of the most important operations in the model. Row-wise attention means that, for each sequence in the MSA, attention runs along the row: residues of the same sequence attend to each other. The “with pair bias” part means that information from is injected into the attention weights as a learned bias term.
Concretely, for a sequence , the attention weight between residues and is:
where and are query and key projections of (sequence , residue , head ), and is a per-head bias term computed linearly from . Note that there’s no subscript on the bias — it’s shared across all sequences in the MSA, because the pair representation is about the target, not about any one alignment row.
class MSARowAttentionWithPairBias(torch.nn.Module):
"""MSA row-wise gated self-attention with pair bias (Algorithm 7).
For each MSA row s, standard multi-head self-attention over residues
``i, j``, with the pair representation z injected as a learned
per-head bias: ``a_{sij}^h = softmax_j(q · k / sqrt(c) + b_{ij}^h)``
where ``b_{ij}^h = LinearNoBias(LayerNorm(z_{ij}))`` (line 3). The
output is gated by ``sigmoid(Linear(m)) ⊙ attention_output`` and
projected back to ``c_m``. The pair bias is what lets the pair rep
influence the MSA rep inside a single Evoformer block — the
symmetric path through :class:`OuterProductMean` fires on the way
back in step 4.
"""
def __init__(self, config):
super().__init__()
self.layer_norm_msa = torch.nn.LayerNorm(config.c_m)
self.layer_norm_pair = torch.nn.LayerNorm(config.c_z)
self.head_dim = config.dim
self.num_heads = config.num_heads
self.total_dim = self.head_dim * self.num_heads
self.linear_q = torch.nn.Linear(in_features=config.c_m, out_features=self.total_dim, bias=False)
self.linear_k = torch.nn.Linear(in_features=config.c_m, out_features=self.total_dim, bias=False)
self.linear_v = torch.nn.Linear(in_features=config.c_m, out_features=self.total_dim, bias=False)
self.linear_pair = torch.nn.Linear(in_features=config.c_z, out_features=self.num_heads, bias=False)
self.linear_gate = torch.nn.Linear(in_features=config.c_m, out_features=self.total_dim)
self.linear_output = torch.nn.Linear(in_features=self.total_dim, out_features=config.c_m)
init_linear(self.linear_q, init="default")
init_linear(self.linear_k, init="default")
init_linear(self.linear_v, init="default")
init_linear(self.linear_pair, init="default")
init_gate_linear(self.linear_gate)
init_linear(self.linear_output, init="final")
def forward(self, msa_representation: torch.Tensor, pair_representation: torch.Tensor,
msa_mask: Optional[torch.Tensor] = None):
msa_representation = self.layer_norm_msa(msa_representation)
# Shape (batch, N_seq, N_res, self.total_dim)
Q = self.linear_q(msa_representation)
K = self.linear_k(msa_representation)
V = self.linear_v(msa_representation)
# Reshape to (batch, N_seq, N_res, self.num_heads, self.head_dim)
Q = Q.reshape((Q.shape[0], Q.shape[1], Q.shape[2], self.num_heads, self.head_dim))
K = K.reshape((K.shape[0], K.shape[1], K.shape[2], self.num_heads, self.head_dim))
V = V.reshape((V.shape[0], V.shape[1], V.shape[2], self.num_heads, self.head_dim))
G = self.linear_gate(msa_representation)
G = G.reshape((G.shape[0], G.shape[1], G.shape[2], self.num_heads, self.head_dim))
# Squash values in range 0 to 1 to act as gating mechanism
G = torch.sigmoid(G)
pair_representation = self.layer_norm_pair(pair_representation)
# Algorithm 7 line 3: b_ij^h = LinearNoBias(LayerNorm(z_ij)).
# Shape (batch, N_res, N_res, self.num_heads)
B = self.linear_pair(pair_representation)
# Align B's axes with the score tensor below: (batch, num_heads, i, j),
# then broadcast across the MSA sequence dim.
B = B.permute(0, 3, 1, 2) # (batch, num_heads, N_res_i, N_res_j)
B = B.unsqueeze(1) # (batch, 1, num_heads, N_res_i, N_res_j)
# Algorithm 7 line 5: a_sij^h = softmax_j(1/sqrt(c) q_si^h . k_sj^h + b_ij^h)
# scores shape: (batch, N_seq, num_heads, N_res_i, N_res_j)
scores = torch.einsum('bsihd, bsjhd -> bshij', Q, K)
scores = scores / math.sqrt(self.head_dim) + B
# Apply MSA mask to key positions (j dimension)
if msa_mask is not None:
# msa_mask: (batch, N_seq, N_res) -> (batch, N_seq, 1, 1, N_res)
mask_bias = (1.0 - msa_mask[:, :, None, None, :]) * (-1e9)
scores = scores + mask_bias
attention = torch.nn.functional.softmax(scores, dim=-1)
# Shape (batch, N_seq, N_res, self.num_heads, self.head_dim)
values = torch.einsum('bshij, bsjhd -> bsihd', attention, V)
values = G * values
values = values.reshape((Q.shape[0], Q.shape[1], Q.shape[2], -1))
output = self.linear_output(values)
# Zero out padded query positions
if msa_mask is not None:
output = output * msa_mask[..., None]
return outputHere is the architectural trick: in the main Evoformer stack, the pair representation enters the MSA stack through this bias. That is the communication channel from back into . Without the bias, each row of the MSA would be processed mostly on its own terms, and the model would have a much weaker way to use current pairwise beliefs to guide residue-level attention. With the bias, every head gets a per-pair signal saying, in effect, “pay attention to positions and together in proportion to what we currently believe about their relationship.” As the Evoformer iterates and accumulates structural information through earlier outer product mean updates, triangle updates, and recycling, the bias becomes more informative, and row attention becomes progressively more structure-aware.
The gating term, a sigmoid on a learned linear projection, matters too, though it is easy to breeze past. The gate lets each head decide how much of its proposed update should actually be written back into the residual stream. The paper does not isolate gating as its own ablation, but the axial-attention ablation removes triangles, pair bias, and gating together and pays a real accuracy cost. The broader point is that these gates are part of the Evoformer’s stabilization machinery rather than decorative notation.
5. Column attention, transition, and the outer product mean
Row attention teaches each sequence about its own residues. The next three sub-blocks do the complementary work: they let the MSA reason across sequences, shape the representation with a transition MLP, and then push the aggregated evolutionary signal into so the pair stack can use it.
Before going through the sub-blocks, the distinction between row- and column-wise attention is worth seeing visually. Both operate on the same MSA tensor; they just run along perpendicular axes. Toggle between them in the widget below. The scope of a single attention step is either one organism’s whole chain (rows) or one aligned position across every organism (columns).
Row-wise attention. Within one sequence, every residue attends to every other residue in the same sequence — attention runs along the amino-acid chain. No information crosses between organisms in this step.
5.1 MSA column-wise attention (Algorithm 8)
Column attention is the dual of row attention. For each residue position , attention runs down the column, and sequences at the same aligned position attend to each other. That is, for residue in sequence , the embedding gets updated using information from residue in sequences .
class MSAColumnAttention(torch.nn.Module):
"""MSA column-wise gated self-attention (Algorithm 8).
For each residue column i, attend across MSA sequences
``s = 1, ..., N_seq`` with standard multi-head attention on ``m_{si}``
(no pair bias, unlike the row variant). Gated by
``sigmoid(Linear(m))`` and projected back to ``c_m``. No dropout
per Algorithm 6. Used only in the main Evoformer; the extra MSA
stack uses :class:`MSAColumnGlobalAttention` instead.
"""
def __init__(self, config):
super().__init__()
self.layer_norm_msa = torch.nn.LayerNorm(config.c_m)
self.head_dim = config.dim
self.num_heads = config.num_heads
self.total_dim = self.head_dim * self.num_heads
self.linear_q = torch.nn.Linear(in_features=config.c_m, out_features=self.total_dim, bias=False)
self.linear_k = torch.nn.Linear(in_features=config.c_m, out_features=self.total_dim, bias=False)
self.linear_v = torch.nn.Linear(in_features=config.c_m, out_features=self.total_dim, bias=False)
self.linear_gate = torch.nn.Linear(in_features=config.c_m, out_features=self.total_dim)
self.linear_output = torch.nn.Linear(in_features=self.total_dim, out_features=config.c_m)
init_linear(self.linear_q, init="default")
init_linear(self.linear_k, init="default")
init_linear(self.linear_v, init="default")
init_gate_linear(self.linear_gate)
init_linear(self.linear_output, init="final")
def forward(self, msa_representation: torch.Tensor, msa_mask: Optional[torch.Tensor] = None):
msa_representation = self.layer_norm_msa(msa_representation)
# Shape (batch, N_seq, N_res, self.total_dim)
Q = self.linear_q(msa_representation)
K = self.linear_k(msa_representation)
V = self.linear_v(msa_representation)
# Reshape to (batch, N_seq, N_res, self.num_heads, self.head_dim)
Q = Q.reshape((Q.shape[0], Q.shape[1], Q.shape[2], self.num_heads, self.head_dim))
K = K.reshape((K.shape[0], K.shape[1], K.shape[2], self.num_heads, self.head_dim))
V = V.reshape((V.shape[0], V.shape[1], V.shape[2], self.num_heads, self.head_dim))
G = self.linear_gate(msa_representation)
G = G.reshape((G.shape[0], G.shape[1], G.shape[2], self.num_heads, self.head_dim))
# Squash values in range 0 to 1 to act as gating mechanism
G = torch.sigmoid(G)
# Shape (batch, N_res, self.num_heads, N_seq, N_seq)
scores = torch.einsum('bsihd, btihd -> bihst', Q, K)
scores = scores / math.sqrt(self.head_dim)
# Apply MSA mask to key positions (t dimension = sequences)
if msa_mask is not None:
# msa_mask: (batch, N_seq, N_res) -> (batch, N_res, 1, 1, N_seq)
mask_bias = (1.0 - msa_mask.permute(0, 2, 1)[:, :, None, None, :]) * (-1e9)
scores = scores + mask_bias
attention = torch.nn.functional.softmax(scores, dim=-1)
# Shape (batch, N_seq, N_res, self.num_heads, self.head_dim)
values = torch.einsum('bihst, btihd -> bsihd', attention, V)
values = G * values
values = values.reshape((Q.shape[0], Q.shape[1], Q.shape[2], -1))
output = self.linear_output(values)
# Zero out padded query positions
if msa_mask is not None:
output = output * msa_mask[..., None]
return outputThere is no pair bias here. The pair representation is a residue-pair object, while column attention is happening across sequences at the same residue position. This is essentially gated multi-head attention along the axis rather than the axis. Column attention lets the model ask, “what does residue 47 look like across this whole family of homologs?” and compress the answer into the target row’s representation.
5.2 MSA transition (Algorithm 9)
A standard two-layer MLP applied per-residue, per-sequence. The hidden dimension is 4× the input dimension — the same 4× expansion pattern as a Transformer FFN — and the nonlinearity is ReLU.
class MSATransition(torch.nn.Module):
"""MSA transition (Algorithm 9).
Two-layer feed-forward applied per MSA cell: ``LayerNorm → Linear(c
→ n·c) → ReLU → Linear(n·c → c)`` with ``n = 4`` by default. The
widening factor ``n`` is the supplement's ``n`` parameter, kept
configurable so the extra MSA stack can choose its own (``c_in``
also overrideable so the same module serves both ``c_m`` and
``c_e`` MSA reps). No dropout — the residual connection is purely
additive per Algorithm 6.
"""
def __init__(self, config, c_in: Optional[int] = None, n: Optional[int] = None):
super().__init__()
self.c_in = config.c_m if c_in is None else c_in
self.n = config.msa_transition_n if n is None else n
self.layer_norm = torch.nn.LayerNorm(self.c_in)
self.linear_up = torch.nn.Linear(in_features=self.c_in, out_features=self.n * self.c_in)
self.linear_down = torch.nn.Linear(in_features=self.c_in * self.n, out_features=self.c_in)
init_linear(self.linear_up, init="relu")
init_linear(self.linear_down, init="final")
def forward(self, msa_representation: torch.Tensor):
msa_representation = self.layer_norm(msa_representation)
activations = self.linear_up(msa_representation)
return self.linear_down(torch.nn.functional.relu(activations))Nothing surprising happens here. The transition is the MSA stack’s feed-forward computation step, analogous to the FFN in a standard Transformer block. It gives the network capacity to combine the features that row and column attention have just surfaced.
5.3 The outer product mean (Algorithm 10)
And now, the payoff. The outer product mean takes the MSA representation, which by this point in the block has been updated by row attention, column attention, and the transition MLP, and converts it into an update for the pair representation. Its job is to take everything the model has learned about residues across the aligned sequences and distill it into a signal about each residue pair in the target sequence. That signal is then added into , where the pair stack can use it to reason about contacts.
class OuterProductMean(torch.nn.Module):
"""Outer product mean (Algorithm 10).
Symmetric MSA → pair update: project each MSA cell to two hidden
vectors ``a_{si}, b_{si} ∈ R^{c_hidden}``, take the MSA mean of
their outer product ``mean_s (a_{si} ⊗ b_{sj})``, flatten to
``c_hidden^2`` channels, and project back to ``c_z``. This is the
only channel in the Evoformer where the MSA rep writes into the
pair rep; the reverse direction (pair → MSA) goes through the
pair-biased row attention.
``c_in``/``c_hidden`` are configurable so the extra MSA stack can
run a narrower OPM.
"""
def __init__(self, config, c_in: Optional[int] = None, c_hidden: Optional[int] = None):
super().__init__()
self.c_in = config.c_m if c_in is None else c_in
self.layer_norm = torch.nn.LayerNorm(self.c_in)
self.c = config.outer_product_dim if c_hidden is None else c_hidden
self.linear_left = torch.nn.Linear(self.c_in, self.c)
self.linear_right = torch.nn.Linear(self.c_in, self.c)
self.linear_out = torch.nn.Linear(in_features=self.c*self.c, out_features=config.c_z)
init_linear(self.linear_left, init="default")
init_linear(self.linear_right, init="default")
init_linear(self.linear_out, init="final")
def forward(self, msa_representation: torch.Tensor, msa_mask: Optional[torch.Tensor] = None):
# msa_mask: (batch, N_seq, N_res) — 1 for valid, 0 for padding
msa_representation = self.layer_norm(msa_representation)
# Shape (batch, N_seq, N_res, self.c)
A = self.linear_left(msa_representation)
B = self.linear_right(msa_representation)
if msa_mask is not None:
# Zero out padded MSA rows before outer product
m = msa_mask.to(A.dtype) # (batch, N_seq, N_res)
A = A * m[..., None] # (batch, N_seq, N_res, c)
B = B * m[..., None]
# Sum over N_seq: (batch, N_res_i, N_res_j, c, c)
outer = torch.einsum('bsic, bsjd -> bijcd', A, B)
if msa_mask is not None:
# Mask-aware normalization: count valid (s,i)*(s,j) pairs
m = msa_mask.to(A.dtype)
norm = torch.einsum('bsi, bsj -> bij', m, m).clamp(min=1.0) # (batch, N_res, N_res)
mean_val = outer / norm[..., None, None]
else:
mean_val = outer / msa_representation.shape[1]
# Shape (batch, N_res, N_res, self.c*self.c)
mean_val = mean_val.reshape(mean_val.shape[0], mean_val.shape[1], mean_val.shape[2], -1)
return self.linear_out(mean_val)The mechanics work as follows. We start from the MSA representation of shape . After a LayerNorm, we apply two separate linear projections to produce a “left” tensor and a “right” tensor , each of shape , where is a smaller hidden dimension. You can think of as the representation of residue “as a left-hand partner” and as the representation of residue “as a right-hand partner” — two different views of the same residue, each tuned for its role in the pairwise interaction.
Then comes the core operation. For every sequence in the MSA and every pair of residue positions , we form the outer product of and . That outer product is a matrix capturing every pairwise interaction between the features of residue and the features of residue , as seen in sequence . A dot product would collapse this relationship down to a single number. The outer product preserves the full feature-by-feature interaction grid.
We then sum these outer products over all sequences in the MSA and divide by the number of valid sequences, with masking used to ignore padded rows. This gives us, for each pair , the average feature-by-feature interaction between residue and residue across the entire alignment. Intuitively, if residues and consistently co-vary across the MSA, the hallmark of co-evolution and likely physical contact, that pattern should show up consistently in these outer products and survive the averaging. If they vary independently, the signals across sequences should wash each other out.
The resulting tensor has shape . We flatten the last two dimensions to get , then apply a final linear projection to map down to , the channel dimension of the pair representation. That produces a tensor of shape that can be added directly into .
The whole process is easier to understand visually than verbally. The widget below walks through a toy MSA (6 sequences, ) with a fixed target pair . Watch the outer-product matrix for each sequence enter the running mean. Patterns shared across sequences reinforce, idiosyncratic patterns wash out, and what survives is the per-pair signal the pair stack will build on top of.
This is the cleanest handoff in the whole architecture. The MSA stack uses row- and column-wise attention to figure out which residues behave similarly across evolutionary history, and the outer product mean then translates those cross-sequence patterns into a per-pair signal. It tells the pair representation, for every , how strongly the evolutionary record suggests these two residues are coupled. The Structure Module downstream uses this — routed through the triangle updates and the IPA in the Structure Module — to guess which residues are in contact, which is what ultimately drives accurate structure prediction.
Putting it all together: the MSA steps compute a representation that optimally captures similarity of residues within sequences (via row attention) and across sequences (via column attention). The outer product mean then uses that representation to generate a measure of similarity between every pair of residues in the target and adds it into . In essence, we use the MSA to figure out which residues are similar to which other residues, and then hand this information to the pair stack so the Structure Module can guess at which residues are in contact.
6. Triangle multiplicative updates
After the MSA stack has finished — row attention, column attention, transition, and the outer product mean — the pair representation holds a first pass at the evolutionary signal. For every residue pair in the target, is a vector encoding what the model currently believes about that pair. The rest of the Evoformer block is about turning this pairwise signal into something geometrically coherent enough for the Structure Module to use. The first two sub-blocks that do this are the triangle multiplicative updates.
The intuition lives in the word “triangle.” Think of as a directed graph on residues. Residues are vertices, and the pair representation is the vector sitting on the directed edge from to . Physical contacts imply consistency constraints over these edges. If residue and residue are close, and residue and residue are close, then residue and residue are also more likely to be close, because all three residues may occupy the same local region of 3D space. If you update every edge independently, the pair representation can tell a globally incoherent story in which each edge is locally plausible but the triangles do not close.
The triangle multiplicative update is designed to address precisely this issue. For each target edge , it computes an update by summing over every residue , multiplying together the other two edges of a triangle :
That’s the outgoing variant. Two linear projections of produce and — you can think of as “the view of the edge that is being used to update outgoing edges at ” and as “the view of the edge for the same purpose.” For each , the elementwise product combines these two views. We then sum over every , linearly project back to pair-rep dimension, gate with a learned sigmoid , and add into .
The incoming variant is the symmetric move, operating on columns instead of rows:
Same operation, different triangle. In outgoing, we’re using the edges emanating from and from to ; in incoming, we’re using the edges arriving at and at from . Together, they cover both directional structures the pair representation is asymmetric in.
6.1 Seeing the operation
Before we look at code, the update is much easier to understand when you can watch a single -sweep fire. The widget below draws the pair representation as an grid. Click any cell to pick the target pair , then watch the animation sum over . In outgoing mode, the two contributing cells sit on row and row ; in incoming mode, they sit on column and column . The dashed triangle connects the three cells in play at the current step of the sum.
z_{2,7} += a_{2,0} ⊙ b_{7,0}A few things are worth noticing as the animation runs. First, the target cell stays fixed, but the two contributing cells sweep across the grid together as advances. Second, in outgoing mode, the two contributing cells share a column (); in incoming mode, they share a row. Third, the -summation touches every other residue in the chain, not just residues currently believed to be nearby. This is a dense update, not a sparse attention pattern.
6.2 The code
With the operation in mind, the PyTorch follows directly. Here is the outgoing variant:
class TriangleMultiplicationOutgoing(torch.nn.Module):
"""Triangle multiplicative update, outgoing edges (Algorithm 11).
Update ``z_ij`` from the two *outgoing* edges of the triangle
``(i, j, k)``: ``z_ij ← g_ij ⊙ Linear(LayerNorm(sum_k a_ik ⊙
b_jk))`` where ``a = gate_a ⊙ projection_a(z)`` and likewise for
``b``. Enforces the triangle-inequality structure across the pair
rep. Algorithm 11 pools over intermediate node ``k`` via the
outgoing edges ``z_{ik}`` and ``z_{jk}``; the incoming-edge
counterpart is :class:`TriangleMultiplicationIncoming`.
"""
def __init__(self, config, c: Optional[int] = None):
super().__init__()
mult_c = config.triangle_mult_c if c is None else c
self.layer_norm_pair = torch.nn.LayerNorm(config.c_z)
self.layer_norm_out = torch.nn.LayerNorm(mult_c)
self.gate1 = torch.nn.Linear(in_features=config.c_z, out_features=mult_c)
self.gate2 = torch.nn.Linear(in_features=config.c_z, out_features=mult_c)
self.linear1 = torch.nn.Linear(in_features=config.c_z, out_features=mult_c)
self.linear2 = torch.nn.Linear(in_features=config.c_z, out_features=mult_c)
self.gate = torch.nn.Linear(in_features=config.c_z, out_features=config.c_z)
self.out_linear = torch.nn.Linear(in_features=mult_c, out_features=config.c_z)
init_gate_linear(self.gate1)
init_gate_linear(self.gate2)
init_linear(self.linear1, init="default")
init_linear(self.linear2, init="default")
init_gate_linear(self.gate)
init_linear(self.out_linear, init="final")
def forward(self, pair_representation: torch.Tensor, pair_mask: Optional[torch.Tensor] = None):
pair_representation = self.layer_norm_pair(pair_representation)
# Shape (batch, N_res, N_res, c)
A = torch.sigmoid(self.gate1(pair_representation)) * self.linear1(pair_representation)
B = torch.sigmoid(self.gate2(pair_representation)) * self.linear2(pair_representation)
# Mask out padded positions before contraction
if pair_mask is not None:
A = A * pair_mask[..., None]
B = B * pair_mask[..., None]
# Shape (batch, N_res, N_res, c_z)
G = torch.sigmoid(self.gate(pair_representation))
# A: (batch, N_res_i, N_res_k, c)
# B: (batch, N_res_j, N_res_k, c)
# Result: (batch, N_res_i, N_res_j, c)
vals = torch.einsum('bikc, bjkc -> bijc', A, B)
# Shape (batch, N_res, N_res, c_z)
out = G * self.out_linear(self.layer_norm_out(vals))
if pair_mask is not None:
out = out * pair_mask[..., None]
return outThe pattern is exactly what the equation says: LayerNorm, four linear projections (linear_a, linear_b, plus gate linears), compute and , einsum the sum over , LayerNorm, final gated linear, done. The einsum string is where the operation lives: bikc,bjkc->bijc says “for each batch , sum over the elementwise product of and , producing an update for in channels .” Read it once carefully; this single einsum is the whole triangle multiplicative update.
The incoming variant differs only in the einsum indices:
class TriangleMultiplicationIncoming(torch.nn.Module):
"""Triangle multiplicative update, incoming edges (Algorithm 12).
Symmetric partner of :class:`TriangleMultiplicationOutgoing`: pool
over intermediate node ``k`` using the *incoming* edges
``z_{ki}`` and ``z_{kj}`` (i.e. ``sum_k a_ki ⊙ b_kj``). Outgoing and
incoming variants fire back-to-back in every Evoformer block so the
pair rep sees both triangle orientations per iteration.
"""
def __init__(self, config, c: Optional[int] = None):
super().__init__()
mult_c = config.triangle_mult_c if c is None else c
self.layer_norm_pair = torch.nn.LayerNorm(config.c_z)
self.layer_norm_out = torch.nn.LayerNorm(mult_c)
self.gate1 = torch.nn.Linear(in_features=config.c_z, out_features=mult_c)
self.gate2 = torch.nn.Linear(in_features=config.c_z, out_features=mult_c)
self.linear1 = torch.nn.Linear(in_features=config.c_z, out_features=mult_c)
self.linear2 = torch.nn.Linear(in_features=config.c_z, out_features=mult_c)
self.gate = torch.nn.Linear(in_features=config.c_z, out_features=config.c_z)
self.out_linear = torch.nn.Linear(in_features=mult_c, out_features=config.c_z)
init_gate_linear(self.gate1)
init_gate_linear(self.gate2)
init_linear(self.linear1, init="default")
init_linear(self.linear2, init="default")
init_gate_linear(self.gate)
init_linear(self.out_linear, init="final")
def forward(self, pair_representation: torch.Tensor, pair_mask: Optional[torch.Tensor] = None):
pair_representation = self.layer_norm_pair(pair_representation)
# Shape (batch, N_res, N_res, c)
A = torch.sigmoid(self.gate1(pair_representation)) * self.linear1(pair_representation)
B = torch.sigmoid(self.gate2(pair_representation)) * self.linear2(pair_representation)
# Mask out padded positions before contraction
if pair_mask is not None:
A = A * pair_mask[..., None]
B = B * pair_mask[..., None]
# Shape (batch, N_res, N_res, c_z)
G = torch.sigmoid(self.gate(pair_representation))
# A: (batch, N_res_k, N_res_i, c)
# B: (batch, N_res_k, N_res_j, c)
# Result: (batch, N_res_i, N_res_j, c)
vals = torch.einsum('bkic, bkjc -> bijc', A, B)
# Shape (batch, N_res, N_res, c_z)
out = G * self.out_linear(self.layer_norm_out(vals))
if pair_mask is not None:
out = out * pair_mask[..., None]
return outbkic,bkjc->bijc: swap which axis sums over. Everything else is identical.
It’s worth pausing on the cost of this operation. The einsum is per call — for a protein of length at channel width , that’s roughly 2 billion multiply-adds per triangle update, and the Evoformer applies two of them (outgoing and incoming) per block, across 48 blocks, up to 4 recycling cycles. This is the single most expensive operation in AlphaFold2’s forward pass, and it’s the reason training and inference both benefit dramatically from gradient checkpointing (which minAlphaFold applies around the Evoformer stack). Without it, the activation memory alone would blow out even an A100.
7. Triangle self-attention
The triangle multiplicative updates enforce triangle consistency in a direct algebraic way: every edge is refined using a sum over third vertices. The triangle self-attention layers are the softer, attention-weighted version of the same idea. They let each edge attend to other edges that share a residue with it, using learned compatibility scores that also reflect the current pair representation.
There are two variants again, differing in how the “shared residue” is picked.
Starting-node attention (Algorithm 13). For each target edge , the query comes from , and the keys and values come from the set of edges starting at , namely . The attention scores also get a bias term derived from , so the third edge of the triangle enters the attention pattern directly.
class TriangleAttentionStartingNode(torch.nn.Module):
"""Triangle self-attention around the starting node (Algorithm 13).
Gated multi-head self-attention over the pair rep with a
triangle-consistency bias: for fixed starting node i, attend over
ending nodes j with keys from ``z_{ij}`` and values from
``z_{ik}``, plus a pair bias ``b_{jk} = LinearNoBias(LayerNorm(
z_{jk}))``. Row-wise dropout (supplement 1.11.6) matches Algorithm 6.
"""
def __init__(self, config, c: Optional[int] = None, num_heads: Optional[int] = None):
super().__init__()
self.layer_norm = torch.nn.LayerNorm(config.c_z)
self.head_dim = config.triangle_dim if c is None else c
self.num_heads = config.triangle_num_heads if num_heads is None else num_heads
self.total_dim = self.head_dim * self.num_heads
self.linear_q = torch.nn.Linear(in_features=config.c_z, out_features=self.total_dim, bias=False)
self.linear_k = torch.nn.Linear(in_features=config.c_z, out_features=self.total_dim, bias=False)
self.linear_v = torch.nn.Linear(in_features=config.c_z, out_features=self.total_dim, bias=False)
self.linear_bias = torch.nn.Linear(in_features=config.c_z, out_features=self.num_heads, bias=False)
self.linear_gate = torch.nn.Linear(in_features=config.c_z, out_features=self.total_dim)
self.linear_output = torch.nn.Linear(in_features=self.total_dim, out_features=config.c_z)
init_linear(self.linear_q, init="default")
init_linear(self.linear_k, init="default")
init_linear(self.linear_v, init="default")
init_linear(self.linear_bias, init="default")
init_gate_linear(self.linear_gate)
init_linear(self.linear_output, init="final")
def forward(self, pair_representation: torch.Tensor, pair_mask: Optional[torch.Tensor] = None):
pair_representation = self.layer_norm(pair_representation)
# Shape (batch, N_res, N_res, self.total_dim)
Q = self.linear_q(pair_representation)
K = self.linear_k(pair_representation)
V = self.linear_v(pair_representation)
# Reshape to (batch, N_res, N_res, self.num_heads, self.head_dim)
Q = Q.reshape((Q.shape[0], Q.shape[1], Q.shape[2], self.num_heads, self.head_dim))
K = K.reshape((K.shape[0], K.shape[1], K.shape[2], self.num_heads, self.head_dim))
V = V.reshape((V.shape[0], V.shape[1], V.shape[2], self.num_heads, self.head_dim))
G = self.linear_gate(pair_representation)
G = G.reshape((G.shape[0], G.shape[1], G.shape[2], self.num_heads, self.head_dim))
# Squash values in range 0 to 1 to act as gating mechanism
G = torch.sigmoid(G)
# Shape (batch, N_res, N_res, self.num_heads)
B = self.linear_bias(pair_representation)
# Q shape (batch, N_res_i, N_res_j, self.num_heads, self.head_dim)
# K shape (batch, N_res_i, N_res_k, self.num_heads, self.head_dim)
# B shape (batch, N_res_j, N_res_k, self.num_heads)
# Output shape (batch, N_res_i, N_res_j, N_res_k, self.num_heads)
scores = torch.einsum('bijhd, bikhd -> bijkh', Q, K)
scores = scores / math.sqrt(self.head_dim) + B.unsqueeze(1)
# Apply pair mask to key positions (k dimension, for a given i)
if pair_mask is not None:
# pair_mask: (batch, N_res, N_res) -> (batch, N_res_i, 1, N_res_k, 1)
mask_bias = (1.0 - pair_mask[:, :, None, :, None]) * (-1e9)
scores = scores + mask_bias
attention = torch.nn.functional.softmax(scores, dim=3)
# Shape (batch, N_res, N_res, self.num_heads, self.head_dim)
values = torch.einsum('bijkh, bikhd -> bijhd', attention, V)
values = G * values
values = values.reshape((Q.shape[0], Q.shape[1], Q.shape[2], -1))
output = self.linear_output(values)
# Zero out padded query positions
if pair_mask is not None:
output = output * pair_mask[..., None]
return outputEnding-node attention (Algorithm 14). The same thing, but now the keys and values come from the edges ending at , namely , and the bias comes from .
class TriangleAttentionEndingNode(torch.nn.Module):
"""Triangle self-attention around the ending node (Algorithm 14).
Mirror image of :class:`TriangleAttentionStartingNode`: fix the
ending node j and attend over starting nodes i. The pair bias is
``b_{ki} = LinearNoBias(LayerNorm(z_{ki}))``. The supplement
prescribes column-wise dropout (not row-wise) on this output — the
Evoformer block applies it accordingly.
"""
def __init__(self, config, c: Optional[int] = None, num_heads: Optional[int] = None):
super().__init__()
self.layer_norm = torch.nn.LayerNorm(config.c_z)
self.head_dim = config.triangle_dim if c is None else c
self.num_heads = config.triangle_num_heads if num_heads is None else num_heads
self.total_dim = self.head_dim * self.num_heads
self.linear_q = torch.nn.Linear(in_features=config.c_z, out_features=self.total_dim, bias=False)
self.linear_k = torch.nn.Linear(in_features=config.c_z, out_features=self.total_dim, bias=False)
self.linear_v = torch.nn.Linear(in_features=config.c_z, out_features=self.total_dim, bias=False)
self.linear_bias = torch.nn.Linear(in_features=config.c_z, out_features=self.num_heads, bias=False)
self.linear_gate = torch.nn.Linear(in_features=config.c_z, out_features=self.total_dim)
self.linear_output = torch.nn.Linear(in_features=self.total_dim, out_features=config.c_z)
init_linear(self.linear_q, init="default")
init_linear(self.linear_k, init="default")
init_linear(self.linear_v, init="default")
init_linear(self.linear_bias, init="default")
init_gate_linear(self.linear_gate)
init_linear(self.linear_output, init="final")
def forward(self, pair_representation: torch.Tensor, pair_mask: Optional[torch.Tensor] = None):
pair_representation = self.layer_norm(pair_representation)
# Shape (batch, N_res, N_res, self.total_dim)
Q = self.linear_q(pair_representation)
K = self.linear_k(pair_representation)
V = self.linear_v(pair_representation)
# Reshape to (batch, N_res, N_res, self.num_heads, self.head_dim)
Q = Q.reshape((Q.shape[0], Q.shape[1], Q.shape[2], self.num_heads, self.head_dim))
K = K.reshape((K.shape[0], K.shape[1], K.shape[2], self.num_heads, self.head_dim))
V = V.reshape((V.shape[0], V.shape[1], V.shape[2], self.num_heads, self.head_dim))
G = self.linear_gate(pair_representation)
G = G.reshape((G.shape[0], G.shape[1], G.shape[2], self.num_heads, self.head_dim))
# Squash values in range 0 to 1 to act as gating mechanism
G = torch.sigmoid(G)
# Shape (batch, N_res, N_res, self.num_heads)
B = self.linear_bias(pair_representation)
# Algorithm 14 line 5: a_ijk^h = softmax_k(1/sqrt(c) q_ij^h . k_kj^h + b_ki^h).
# The highlighted differences from the starting-node version (Algorithm 13)
# are that keys/values are indexed (k, j) instead of (i, k), and the bias
# is b_{k,i} instead of b_{j,k}.
#
# Q shape (batch, N_res_i, N_res_j, num_heads, head_dim)
# K shape (batch, N_res_k, N_res_j, num_heads, head_dim)
# B shape (batch, N_res, N_res, num_heads), with B[b, i, j, h] coming from z_{ij}.
# Output shape (batch, N_res_i, N_res_j, N_res_k, num_heads)
scores = torch.einsum('bijhd, bkjhd -> bijkh', Q, K)
# We need B indexed as b^h_{k,i} at score position (b, i, j, k, h), i.e.
# B[b, k, i, h]. Swapping axes 1 and 2 of B yields a view whose indexing
# is B_t[b, x, y, h] = B[b, y, x, h], so B_t[b, i, k, h] = B[b, k, i, h].
# Inserting a new j-axis with unsqueeze(2) broadcasts that bias to every
# (i, j, k, h) score.
scores = scores / math.sqrt(self.head_dim) + B.transpose(1, 2).unsqueeze(2)
# Apply pair mask to key positions: an attention score at (b, i, j, k, h)
# should be masked when the *key* pair z_{k,j} is padding, i.e. when
# pair_mask[b, k, j] == 0. Permute swaps the k/j axes so the view exposes
# [b, j, k], then broadcast over i and h.
if pair_mask is not None:
mask_bias = (1.0 - pair_mask.permute(0, 2, 1)[:, None, :, :, None]) * (-1e9)
scores = scores + mask_bias
attention = torch.nn.functional.softmax(scores, dim=3)
# Shape (batch, N_res, N_res, self.num_heads, self.head_dim)
values = torch.einsum('bijkh, bkjhd -> bijhd', attention, V)
values = G * values
values = values.reshape((Q.shape[0], Q.shape[1], Q.shape[2], -1))
output = self.linear_output(values)
# Zero out padded query positions
if pair_mask is not None:
output = output * pair_mask[..., None]
return outputThe two operations together cover both ways a third residue can be “shared” with the edge : either sits at the start of both and , or sits at the end of both and . The triangle bias, which pulls the third edge of the triangle directly into the softmax, is what makes these triangle attention rather than vanilla row/column attention on the pair representation. Without that bias, the operations would look like ordinary attention over rows and columns of .
After triangle attention, the Evoformer block closes with a simple pair transition MLP — the same 2-layer, 4×-expansion FFN pattern we saw in §5.2, but now on the pair representation:
class PairTransition(torch.nn.Module):
"""Pair transition (Algorithm 15).
Per-pair feed-forward: ``LayerNorm → Linear(c_z → n·c_z) → ReLU →
Linear(n·c_z → c_z)`` with widening factor ``n = 4``. Same shape as
:class:`MSATransition` but over the pair rep instead of the MSA
rep. No dropout per Algorithm 6.
"""
def __init__(self, config, n: Optional[int] = None):
super().__init__()
self.n = config.pair_transition_n if n is None else n
self.layer_norm = torch.nn.LayerNorm(config.c_z)
self.linear_up = torch.nn.Linear(in_features=config.c_z, out_features=self.n*config.c_z)
self.linear_down = torch.nn.Linear(in_features=config.c_z*self.n, out_features=config.c_z)
init_linear(self.linear_up, init="relu")
init_linear(self.linear_down, init="final")
def forward(self, pair_representation: torch.Tensor):
pair_representation = self.layer_norm(pair_representation)
activations = self.linear_up(pair_representation)
return self.linear_down(torch.nn.functional.relu(activations))That is the full Evoformer block: seven sub-blocks, of which three update the MSA, one couples the MSA into the pair representation, and the remainder refine the pair representation. The pair stack is where geometric consistency is enforced, and the block budget reflects that.
8. Extra MSA and templates
Two side pipelines feed additional information into the main representations before the Evoformer trunk runs on them. Both are best understood as cheaper stacks that produce pair-representation updates. Neither gets the full 48-block Evoformer treatment, because neither is meant to carry the main iterative reasoning load.
8.1 Extra MSA
AlphaFold2’s main MSA input is clustered and capped at a manageable number of sequences — enough to carry rich signal, but small enough that the Evoformer’s attention cost does not explode. The extra MSA is a larger slice of the same alignment that gets processed by a shallower, cheaper sibling of the Evoformer: fewer blocks, global column attention instead of full column attention, and a one-way handoff into the pair representation.
class ExtraMsaStack(torch.nn.Module):
"""Extra MSA stack (Algorithm 18, supplement 1.7.2).
Lightweight Evoformer-like block for the unclustered "extra" MSA.
The extra MSA is much deeper (default ``N_extra_seq = 1024`` vs
``N_cluster = 128``) but compressed to a smaller channel dim
``c_e`` to stay cheap. Two differences from the main Evoformer:
* MSA column attention is replaced by
:class:`MSAColumnGlobalAttention` (Algorithm 19) — across
thousands of sequences, per-head K/V sharing is what keeps
the column step tractable.
* Row attention with pair bias is inlined here rather than
reusing :class:`~minalphafold.evoformer.MSARowAttentionWithPairBias`
so ``c_e ≠ c_m`` projections stay self-contained.
Consumes the extra MSA representation and the pair representation;
writes updates back to both (triangle updates + pair transition
apply after the OPM consumes the updated extra MSA).
"""
def __init__(self, config):
super().__init__()
self.layer_norm_msa = torch.nn.LayerNorm(config.c_e)
self.layer_norm_pair = torch.nn.LayerNorm(config.c_z)
self.head_dim = config.extra_msa_dim
self.num_heads = config.num_heads
self.total_dim = self.head_dim * self.num_heads
# MSA row attention with pair bias (inline, same as Algorithm 7)
self.linear_q = torch.nn.Linear(in_features=config.c_e, out_features=self.total_dim, bias=False)
self.linear_k = torch.nn.Linear(in_features=config.c_e, out_features=self.total_dim, bias=False)
self.linear_v = torch.nn.Linear(in_features=config.c_e, out_features=self.total_dim, bias=False)
self.linear_pair = torch.nn.Linear(in_features=config.c_z, out_features=self.num_heads, bias=False)
self.linear_gate = torch.nn.Linear(in_features=config.c_e, out_features=self.total_dim)
self.linear_output = torch.nn.Linear(in_features=self.total_dim, out_features=config.c_e)
init_linear(self.linear_q, init="default")
init_linear(self.linear_k, init="default")
init_linear(self.linear_v, init="default")
init_linear(self.linear_pair, init="default")
init_gate_linear(self.linear_gate)
init_linear(self.linear_output, init="final")
self.msa_col_att = MSAColumnGlobalAttention(config, c_in=config.c_e)
self.msa_transition = MSATransition(
config,
c_in=config.c_e,
n=getattr(config, "extra_msa_transition_n", config.msa_transition_n),
)
self.outer_mean = OuterProductMean(
config,
c_in=config.c_e,
c_hidden=getattr(config, "extra_msa_outer_product_dim", config.outer_product_dim),
)
self.triangle_mult_out = TriangleMultiplicationOutgoing(config)
self.triangle_mult_in = TriangleMultiplicationIncoming(config)
self.triangle_att_start = TriangleAttentionStartingNode(config)
self.triangle_att_end = TriangleAttentionEndingNode(config)
self.pair_transition = PairTransition(config)
self.msa_dropout_p = config.extra_msa_dropout
self.pair_dropout_p = config.extra_pair_dropout
def forward(self, extra_msa_representation: torch.Tensor, pair_representation: torch.Tensor,
extra_msa_mask: Optional[torch.Tensor] = None, pair_mask: Optional[torch.Tensor] = None):
# extra_msa_representation shape: (batch, N_extra_seq, N_res, c_e)
# pair_representation shape: (batch, N_res, N_res, c_z)
# extra_msa_mask: (batch, N_extra_seq, N_res) — 1 for valid, 0 for padding
# pair_mask: (batch, N_res, N_res) — 1 for valid, 0 for padding
msa_representation = self.layer_norm_msa(extra_msa_representation)
pair_norm = self.layer_norm_pair(pair_representation)
# --- MSA row attention with pair bias ---
# Shape (batch, N_extra_seq, N_res, total_dim)
Q = self.linear_q(msa_representation)
K = self.linear_k(msa_representation)
V = self.linear_v(msa_representation)
# Reshape to (batch, N_extra_seq, N_res, num_heads, head_dim)
Q = Q.reshape((Q.shape[0], Q.shape[1], Q.shape[2], self.num_heads, self.head_dim))
K = K.reshape((K.shape[0], K.shape[1], K.shape[2], self.num_heads, self.head_dim))
V = V.reshape((V.shape[0], V.shape[1], V.shape[2], self.num_heads, self.head_dim))
G = self.linear_gate(msa_representation)
G = G.reshape((G.shape[0], G.shape[1], G.shape[2], self.num_heads, self.head_dim))
# Squash values in range 0 to 1 to act as gating mechanism
G = torch.sigmoid(G)
# Pair bias: project pair representation to per-head bias
# Shape (batch, N_res, N_res, num_heads) -> (batch, num_heads, N_res, N_res)
B = self.linear_pair(pair_norm)
B = B.permute(0, 3, 1, 2)
# Add sequence dim for broadcast: (batch, 1, num_heads, N_res, N_res)
B = B.unsqueeze(1)
# Q shape (batch, N_extra_seq, N_res_i, num_heads, head_dim)
# K shape (batch, N_extra_seq, N_res_j, num_heads, head_dim)
# Output shape (batch, N_extra_seq, num_heads, N_res, N_res)
scores = torch.einsum('bsihd, bsjhd -> bshij', Q, K)
scores = scores / math.sqrt(self.head_dim) + B
# Apply extra MSA mask to key positions (j dimension)
if extra_msa_mask is not None:
mask_bias = (1.0 - extra_msa_mask[:, :, None, None, :]) * (-1e9)
scores = scores + mask_bias
attention = torch.nn.functional.softmax(scores, dim=-1)
# Shape (batch, N_extra_seq, N_res, num_heads, head_dim)
values = torch.einsum('bshij, bsjhd -> bsihd', attention, V)
values = G * values
# Reshape to (batch, N_extra_seq, N_res, total_dim)
values = values.reshape((values.shape[0], values.shape[1], values.shape[2], -1))
row_update = self.linear_output(values)
# Zero out padded query positions
if extra_msa_mask is not None:
row_update = row_update * extra_msa_mask[..., None]
# --- MSA representation updates ---
extra_msa_representation = extra_msa_representation + dropout_rowwise(
row_update,
p=self.msa_dropout_p,
training=self.training,
)
extra_msa_representation = extra_msa_representation + self.msa_col_att(
extra_msa_representation, msa_mask=extra_msa_mask)
extra_msa_representation = extra_msa_representation + self.msa_transition(extra_msa_representation)
# --- Pair representation updates ---
pair_representation = pair_representation + self.outer_mean(
extra_msa_representation, msa_mask=extra_msa_mask)
pair_representation = pair_representation + dropout_rowwise(
self.triangle_mult_out(pair_representation, pair_mask=pair_mask),
p=self.pair_dropout_p,
training=self.training,
)
pair_representation = pair_representation + dropout_rowwise(
self.triangle_mult_in(pair_representation, pair_mask=pair_mask),
p=self.pair_dropout_p,
training=self.training,
)
pair_representation = pair_representation + dropout_rowwise(
self.triangle_att_start(pair_representation, pair_mask=pair_mask),
p=self.pair_dropout_p,
training=self.training,
)
pair_representation = pair_representation + dropout_columnwise(
self.triangle_att_end(pair_representation, pair_mask=pair_mask),
p=self.pair_dropout_p,
training=self.training,
)
pair_representation = pair_representation + self.pair_transition(pair_representation)
return extra_msa_representation, pair_representationThe critical substitution is here: where the main MSA uses full column attention, the extra MSA uses MSAColumnGlobalAttention , which computes attention from a pooled summary of each column rather than from all pairwise sequence-to-sequence scores. The broad idea is the same as many efficient-attention methods: trade some expressivity for a much cheaper operation. The extra MSA can afford that tradeoff because its role is not to preserve a richly refined per-sequence representation. Its role is to extract more evolutionary signal and write it into .
8.2 Templates
When AlphaFold2 has structural hints from related proteins in the PDB, it processes them with a dedicated template pair stack. Each template is featurized into a pair representation of shape : distances between backbone atoms, unit vectors between Cα positions, sequence identity masks, and related geometric features. These pair features are then fed through a few Evoformer-style blocks specialized for pair-only updates.
class TemplatePair(torch.nn.Module):
"""Template pair stack (Algorithm 16, supplement 1.7.1).
Per-template shallow Evoformer-like pair stack: each of
``config.template_pair_num_blocks`` blocks applies triangle
self-attention (start + end), triangle multiplication
(outgoing + incoming), and a pair transition. Dropout matches the
supplement — row-wise on starting / multiplicative updates, column-
wise on ending. Batch and template dims are flattened for each
block so templates evolve independently; the final LayerNorm
happens once before the pointwise attention pool in
:class:`TemplatePointwiseAttention`.
"""
def __init__(self, config):
super().__init__()
self.num_blocks = config.template_pair_num_blocks
self.dropout_p = config.template_pair_dropout
# Supplement 1.7.1 / Algorithm 16: the template pair stack overrides
# the main-Evoformer triangle dims (paper default c=64 for both the
# multiplicative and attention updates, and n=2 on the pair transition)
# instead of inheriting triangle_mult_c / triangle_dim / pair_transition_n.
template_tri_mult_c = config.template_triangle_mult_c
template_tri_attn_c = config.template_triangle_attn_c
template_tri_attn_heads = config.template_triangle_attn_num_heads
template_pair_trans_n = config.template_pair_transition_n
self.layer_norm = torch.nn.LayerNorm(config.c_t)
self.linear_in = torch.nn.Linear(in_features=config.c_t, out_features=config.c_z)
init_linear(self.linear_in, init="default")
self.triangle_mult_out = torch.nn.ModuleList(
[TriangleMultiplicationOutgoing(config, c=template_tri_mult_c) for _ in range(self.num_blocks)]
)
self.triangle_mult_in = torch.nn.ModuleList(
[TriangleMultiplicationIncoming(config, c=template_tri_mult_c) for _ in range(self.num_blocks)]
)
self.triangle_att_start = torch.nn.ModuleList(
[
TriangleAttentionStartingNode(config, c=template_tri_attn_c, num_heads=template_tri_attn_heads)
for _ in range(self.num_blocks)
]
)
self.triangle_att_end = torch.nn.ModuleList(
[
TriangleAttentionEndingNode(config, c=template_tri_attn_c, num_heads=template_tri_attn_heads)
for _ in range(self.num_blocks)
]
)
self.pair_transition = torch.nn.ModuleList(
[PairTransition(config, n=template_pair_trans_n) for _ in range(self.num_blocks)]
)
self.final_layer_norm = torch.nn.LayerNorm(config.c_z)
def forward(self, template_feat: torch.Tensor, pair_mask: Optional[torch.Tensor] = None):
# template_feat shape: (batch, N_templates, N_res, N_res, c_t)
# Project from template feature space to pair representation space
# Output shape: (batch, N_templates, N_res, N_res, c_z)
template_feat = self.linear_in(self.layer_norm(template_feat))
b, t, n_i, n_j, c = template_feat.shape
# Merge batch and template dims to process each template independently
# Shape: (batch * N_templates, N_res, N_res, c_z)
pair_representation = template_feat.reshape(b * t, n_i, n_j, c)
flat_pair_mask = None
if pair_mask is not None:
flat_pair_mask = pair_mask.reshape(b * t, n_i, n_j)
pair_representation = pair_representation * flat_pair_mask[..., None]
for block_idx in range(self.num_blocks):
if flat_pair_mask is not None:
pair_representation = pair_representation * flat_pair_mask[..., None]
pair_representation = pair_representation + dropout_rowwise(
self.triangle_att_start[block_idx](pair_representation, pair_mask=flat_pair_mask),
p=self.dropout_p,
training=self.training,
)
pair_representation = pair_representation + dropout_columnwise(
self.triangle_att_end[block_idx](pair_representation, pair_mask=flat_pair_mask),
p=self.dropout_p,
training=self.training,
)
pair_representation = pair_representation + dropout_rowwise(
self.triangle_mult_out[block_idx](pair_representation, pair_mask=flat_pair_mask),
p=self.dropout_p,
training=self.training,
)
pair_representation = pair_representation + dropout_rowwise(
self.triangle_mult_in[block_idx](pair_representation, pair_mask=flat_pair_mask),
p=self.dropout_p,
training=self.training,
)
pair_representation = pair_representation + self.pair_transition[block_idx](pair_representation)
if flat_pair_mask is not None:
pair_representation = pair_representation * flat_pair_mask[..., None]
pair_representation = self.final_layer_norm(pair_representation)
if flat_pair_mask is not None:
pair_representation = pair_representation * flat_pair_mask[..., None]
# Restore batch and template dims
# Output shape: (batch, N_templates, N_res, N_res, c_z)
pair_representation = pair_representation.reshape(b, t, n_i, n_j, c)
return pair_representationThe template pair stack is Evoformer-lite: triangle multiplication (outgoing and incoming), triangle attention (starting and ending), and a pair transition. There is no MSA side. The output of the stack for each template is a pair tensor, and those tensors get combined across templates by a TemplatePointwiseAttention layer that lets the target sequence attend across its available structural hints.
For the minimal implementation and for this walkthrough, templates are present but optional. The
tiny.tomlconfig runs with zero templates; the full config runs with up to four. The important thing to take away is structural — templates enter the pipeline as pair-representation updates, exactly like the OPM output from the main MSA. By the time the Evoformer starts, has already been seeded with (a) relative positional encoding, (b) any template signal, and (c) the extra-MSA contribution. The main MSA will add more in each Evoformer block via the OPM.
9. Recycling
The last structural idea in the Evoformer-level description of AlphaFold2 is recycling. This is one of those ideas that looks almost too simple to be important, but turns out to be load-bearing. The setup is as follows: run a forward pass, get an MSA representation, a pair representation, and a Structure Module output. Then take parts of those outputs, feed them back into the inputs of the next forward pass, and run the whole model again. In the paper setup, this happens up to four times at inference and a sampled number of times during training.
What exactly gets fed back? Three things: the first row of the final MSA representation (, the target row), the final pair representation , and a distance-binned version of the pseudo-Cβ coordinates extracted from the predicted structure. The representation tensors are normalized and added into the current-cycle input embeddings. The pseudo-Cβ distances go through one extra step: recycling_distance_bin one-hot-encodes pairwise distances into 15 radial bins, linearly projects the bins into pair-representation dimension, and adds the result into .
# Algorithm 2 line 6 (= Algorithm 32 / RecyclingEmbedder):
# m_1i += LayerNorm(m_1i^prev)
# z_ij += LayerNorm(z_ij^prev) + Linear(one_hot(d_ij^prev))
# On the first cycle the prev tensors are zero, so these
# additions vanish. Clone before the in-place write to the
# first MSA row so the embedder's output tensor is untouched.
msa_repr = msa_representation.clone()
pair_repr = pair_representation.clone()
msa_repr[:, 0, :, :] += self.recycle_norm_s(single_rep_prev)
pair_repr += self.recycle_norm_z(z_prev)
pair_repr += self.recycle_linear_d(recycling_distance_bin(x_prev, n_bins=15))Two details matter for how this trains.
Stop-gradient between cycles. The tensors fed back are detached from the computation graph. Gradients only flow through the final cycle of a training forward pass; earlier cycles provide refinement but do not receive direct gradient through the recycling loop. This keeps the memory cost much lower than fully unrolling every cycle. It also has a useful regularizing effect: the model has to learn representations that are useful both when consumed by the current cycle’s Structure Module and when consumed by the next cycle’s input embedder.
Random number of cycles during training. At inference, the number of cycles can be fixed. During training, the number of cycles is sampled uniformly. This means the model sees 1-cycle, 2-cycle, and later-cycle inputs, and must produce reasonable predictions in all of them rather than specializing only to the final cycle. Recycling is therefore not just an inference-time refinement trick. It is a training regime that teaches the model to converge iteratively.
I find recycling to be one of the most underrated components of AlphaFold2. It is the mechanism that turns a 48-block Evoformer into an iterative refiner with shared weights. The inference cost rises with the number of cycles, but the paper’s ablations show that removing recycling is one of the more meaningful hits to accuracy. The broader reason is intuitive: the model is trained to consume its own previous predictions, so later cycles operate on inputs that are increasingly close to the distribution the model itself creates.
10. The Structure Module
Everything we have done so far — input embedding, the Evoformer, and recycling — has been aimed at producing two useful representations: a pair representation rich with structural information, and a single representation for each residue of the target sequence. But none of this is yet a protein structure. The model still needs to place atoms in 3D space. The Structure Module is where that happens.
It does this in a way that is genuinely different from the Evoformer. There is no generic self-attention over abstract sequence tensors here. Instead, the core object is the rigid frame, a local coordinate system attached to each residue. The attention weights and coordinate updates are constructed to respect the fact that proteins live in 3D space and do not care how we orient the global axes. That constraint, SE(3) equivariance/invariance depending on the object being discussed, is what gives the Structure Module its distinctive shape.
The module runs for 8 shared-weight iterations. Each iteration performs invariant point attention, a transition MLP, a backbone update, and all-atom coordinate construction. We’ll build up to the full loop in §10e; first, we need to get the geometry straight.
10a. Rigid frames
Every residue in an AlphaFold2 prediction has an associated rigid frame: a translation and a rotation . Together, is an element of the special Euclidean group , the group of rigid motions in 3D. You can think of as a small coordinate system attached to the residue. The origin sits on the residue’s atom; the axis points roughly along the bond; the axis lives in the plane of , , and while remaining orthogonal to ; and the axis is their cross product.
def rigid_frame_from_three_points(
point_on_neg_x_axis: torch.Tensor,
origin: torch.Tensor,
point_on_xy_plane: torch.Tensor,
) -> tuple[torch.Tensor, torch.Tensor]:
"""Gram–Schmidt frame construction (Algorithm 21).
Returns ``(R, t)`` where ``t = origin`` and ``R`` is a 3×3 rotation
matrix whose columns are the local-frame axes expressed in global
coordinates.
Axis conventions for the three inputs:
* ``origin`` becomes the frame centre (translation component).
* ``point_on_neg_x_axis`` sits at **negative** x in the built frame:
the positive x-axis points *from this atom toward the origin*
(``x_axis = (origin − point_on_neg_x_axis) / ||·||``). **This is the
opposite direction from Algorithm 21's ``e1 = (x3 − x2)/||x3 − x2||``**
— when we feed backbone atoms as ``(C, Cα, N)`` below, the backbone
rotation gets reconciled with the paper via
``BACKBONE_FRAME_ADAPTATION``.
* ``point_on_xy_plane`` lies in the xy-plane by construction. Its
residual (after subtracting the x-axis component) defines ±y.
The y/z axes are completed by two cross products so R is guaranteed
orthonormal and right-handed (``det R = +1``).
"""
x_axis = safe_normalize(origin - point_on_neg_x_axis)
xy_axis = point_on_xy_plane - origin
z_axis = safe_normalize(torch.cross(x_axis, xy_axis, dim=-1))
y_axis = safe_normalize(torch.cross(z_axis, x_axis, dim=-1))
rotations = torch.stack([x_axis, y_axis, z_axis], dim=-1)
return rotations, originThe function computes frames from triples of atom coordinates. In AlphaFold2, these are the , , and backbone atoms. The construction is a three-step Gram-Schmidt procedure: translate so is at the origin, orient along , and orient by projecting into the -plane. The specific convention matters because it must match the convention used later to build side chains.
Why carry around frames rather than just Cartesian coordinates? Two reasons. First, frames express geometry relatively. For any two residues and , the position of “as seen by ” is just , meaning residue ‘s translation expressed in residue ‘s local coordinate system. This operation is invariant under any global rigid motion of the whole protein, and that invariance is something we want to build in rather than learn. Second, frames compose cleanly. If you have an update to apply to a residue, the composition is still a valid frame. This is how backbone updates work in §10c, and it is also how side-chain atoms are built by composing torsion-angle rotations with literature frames in §10d.
Quaternions as the rotation parameterization
Rotations have three natural representations: rotation matrices (9 numbers, 6 constraints from orthogonality), Euler angles (3 numbers, but with unpleasant pathologies like gimbal lock and discontinuous wrap-around), and quaternions (4 numbers, 1 constraint from unit norm). AlphaFold2 uses quaternions when a rotation is being output or updated by the model, and rotation matrices when a rotation is being applied.
The reason is mostly about smooth updates. When the Structure Module emits a backbone update, it outputs three scalars, which we can call , and forms an unnormalized quaternion . It then normalizes this quaternion and converts it to a rotation matrix. This parameterization has a useful property: the identity rotation is at , corresponding to . Thus, “no update” is the all-zero model output, which is exactly what we want a small MLP to default to at initialization. Rotation matrices and Euler angles do not give us this nearly as cleanly.
Concretely, a unit quaternion maps to a rotation matrix via:
The widget below is the clearest way I know to internalize this formula. Drag the four sliders to change the quaternion components, and the rotation matrix, the normalized quaternion, and the attached local frame (thick red/green/blue axes, plus a small gray cube) all update live. The world frame stays ghosted in the background for reference.
(1.00, 0.00, 0.00, 0.00)Play with it for a minute. A few things are worth noticing. First, the rotation depends only on the direction of , not its magnitude, because we normalize before converting to a matrix. Second, and give the same rotation matrix; this is the double cover of by the unit quaternions . Third, the presets for “90° about X” (or Y) sit at because quaternions use the half-angle. These details show up constantly when reading rotation code.
def backbone_frames(
atom14_positions: torch.Tensor,
atom14_mask: torch.Tensor,
aatype: torch.Tensor | None = None,The backbone_frames helper just calls rigid_frame_from_three_points in a loop across residues — this is how the ground-truth frames (and, initially, the model’s prior-cycle frames during recycling) get built from atom coordinates.
10b. Invariant Point Attention
Invariant Point Attention is the signature innovation of the Structure Module. Built on top of rigid frames, it defines a three-term attention score that is invariant to any global rigid motion of the input. This is not merely a soft inductive bias. It is an algebraic property of the operation. Rotate or translate the whole protein in space, and the IPA attention scores are unchanged.
The score between a query residue and a key residue for head looks like:
Three terms, three sources of information. The scalar term is a standard attention score on scalar queries and keys, analogous to attention in the Evoformer. The pair bias term injects the current pair representation into the attention pattern, just like row attention did in §4. The new piece is the point distance term.
Each query head carries a small bank of 3D query points . These points are expressed in residue ‘s local frame; applying lifts them into global coordinates. The same is true for the key points . The point-distance term is a negative squared distance between each pair of globally transformed points, summed across points. If a query point lifted from ends up close in 3D to a key point lifted from , that residue pair gets a higher attention score.
Why does this give invariance? Suppose we apply a global rigid motion to every residue. Then and . The transformed points become and . Rigid motions preserve distances, so the squared-distance term is unchanged. The scalar term does not touch positions, and the pair bias comes from , which has no global spatial orientation. Hence, all three terms are preserved, and the attention pattern is preserved.
Here’s what this looks like, conceptually, on a synthetic chain. Click a residue to select it as the query . Top-5 attention weights are drawn as lines from the selected sphere to the others, with thickness proportional to weight, and also listed in the panel on the right. Toggle Auto-rotate scene: the whole chain spins, but the attention pattern stays fixed — individual lines don’t brighten or fade as the scene rotates.
The attention weight for (i → j) depends on Ti−1 · tj— the position of residue j as seen from residue i's local frame. This is invariant under any global rotation of the chain, which is why the connecting lines thicken and fade only when you change the query, not when the scene spins.
The score function in the widget is simplified — I compute a single point per query/key and use a per-residue position only — but the geometric story it tells is the real one. A few things to watch for: (1) when you toggle “Auto-rotate scene,” the view spins but the weights don’t. That’s invariance in action. (2) Attention is not the same as nearest-neighbor in 3D — the sequence-distance penalty gives the pattern an asymmetry that a pure spatial score wouldn’t. (3) When you pick a residue in the middle of the chain, the attention weights are richer than at the ends — another thing a learned attention would mirror.
The code
class InvariantPointAttention(torch.nn.Module):
"""Invariant Point Attention (Algorithm 22, supplement 1.8.2).
The geometry-aware attention that lets the Structure Module reason
about 3-D context while staying equivariant to rigid motions of the
input. For each head, three attention-score contributions are summed:
1. Standard scalar Q·K attention on channel projections of ``s_i``.
2. Pair bias ``b_{ij} = Linear(z_{ij})`` — the pair rep as score bias.
3. "Point" attention on 3-D points ``Q, K, V`` transformed into each
residue's local frame ``T_i``: the attention score for (i, j) is
``-γ_h · ||T_i(q_i^h) - T_j(k_j^h)||^2 / 2`` (line 7). Invariance
to rigid motion of the whole protein is guaranteed because frames
and points move together.
The combined score is ``(1/sqrt(3)) · (scalar + bias + points)`` per
supplement 1.8.2 (line 7), softmax'd over j, and the output pools
scalar values, pair values, and point values (transformed back into
each residue's local frame). ``seq_mask`` zeros both queries and
keys before the softmax so padded residues neither attend nor are
attended to.
"""
def __init__(self, config):
super().__init__()
self.num_heads = config.ipa_num_heads
self.head_dim = config.ipa_c
self.total_dim = self.head_dim * self.num_heads
self.n_query_points = config.ipa_n_query_points
self.n_value_points = config.ipa_n_value_points
self.inf = 1e5
self.eps = 1e-8
# Canonical AF2/OpenFold monomer IPA uses a biased query projection and
# combined key/value projections for both scalar and point features.
self.linear_q = torch.nn.Linear(in_features=config.c_s, out_features=self.total_dim, bias=True)
self.linear_kv = torch.nn.Linear(in_features=config.c_s, out_features=2 * self.total_dim, bias=True)
self.linear_q_points = torch.nn.Linear(
in_features=config.c_s,
out_features=3 * self.num_heads * self.n_query_points,
bias=True,
)
self.linear_kv_points = torch.nn.Linear(
in_features=config.c_s,
out_features=3 * self.num_heads * (self.n_query_points + self.n_value_points),
bias=True,
)
self.linear_bias = torch.nn.Linear(in_features=config.c_z, out_features=self.num_heads, bias=True)
self.linear_output = torch.nn.Linear(
in_features=self.total_dim
+ self.num_heads * self.n_value_points * 4
+ self.num_heads * config.c_z,
out_features=config.c_s,
)
_init_linear(self.linear_q, init="default")
_init_linear(self.linear_kv, init="default")
_init_linear(self.linear_q_points, init="default")
_init_linear(self.linear_kv_points, init="default")
_init_linear(self.linear_bias, init="default")
_init_linear(self.linear_output, init="final")
self.head_weights = torch.nn.Parameter(torch.zeros(self.num_heads))
def _project_points(
self,
linear: torch.nn.Linear,
single_representation: torch.Tensor,
num_points: int,
) -> torch.Tensor:
raw_points = linear(single_representation)
x_coords, y_coords, z_coords = torch.chunk(raw_points, 3, dim=-1)
point_coords = torch.stack([x_coords, y_coords, z_coords], dim=-1)
return point_coords.reshape(
single_representation.shape[0],
single_representation.shape[1],
self.num_heads,
num_points,
3,
)
def _assemble_output_features(
self,
attention: torch.Tensor,
value_scalar: torch.Tensor,
value_points_global: torch.Tensor,
pair_representation: torch.Tensor,
rotations: torch.Tensor,
translation: torch.Tensor,
) -> torch.Tensor:
batch_size = rotations.shape[0]
num_residues = rotations.shape[1]
output_scalar = torch.matmul(
attention,
value_scalar.permute(0, 2, 1, 3).to(dtype=attention.dtype),
).permute(0, 2, 1, 3)
output_scalar = output_scalar.reshape(batch_size, num_residues, -1)
result_point_global = torch.einsum(
"bhij,bjhpc->bihpc",
attention,
value_points_global.to(dtype=attention.dtype),
)
result_point_local = torch.einsum(
"biop,bihqp->bihqo",
rotations.transpose(-1, -2),
result_point_global - translation[:, :, None, None, :],
)
result_point_norms = torch.sqrt(torch.sum(result_point_local ** 2, dim=-1) + self.eps)
result_point_norms = result_point_norms.reshape(batch_size, num_residues, -1)
# Canonical monomer IPA concatenates x/y/z point channels separately.
result_point_local = result_point_local.reshape(batch_size, num_residues, -1, 3)
result_point_x, result_point_y, result_point_z = result_point_local.unbind(dim=-1)
output_pair = torch.einsum(
"bhij,bijd->bihd",
attention,
pair_representation.to(dtype=attention.dtype),
)
output_pair = output_pair.reshape(batch_size, num_residues, -1)
return torch.cat(
[
output_scalar,
result_point_x,
result_point_y,
result_point_z,
result_point_norms,
output_pair,
],
dim=-1,
)
def _forward_output_features(
self,
single_representation: torch.Tensor,
pair_representation: torch.Tensor,
rotations: torch.Tensor,
translation: torch.Tensor,
seq_mask: Optional[torch.Tensor] = None,
) -> torch.Tensor:
batch_size = single_representation.shape[0]
num_residues = single_representation.shape[1]
q = self.linear_q(single_representation).reshape(batch_size, num_residues, self.num_heads, self.head_dim)
kv = self.linear_kv(single_representation).reshape(batch_size, num_residues, self.num_heads, 2 * self.head_dim)
k, v = torch.split(kv, self.head_dim, dim=-1)
q_points = self._project_points(self.linear_q_points, single_representation, self.n_query_points)
kv_points = self._project_points(
self.linear_kv_points,
single_representation,
self.n_query_points + self.n_value_points,
)
k_points, v_points = torch.split(
kv_points,
[self.n_query_points, self.n_value_points],
dim=-2,
)
query_points_global = torch.einsum("biop,bihqp->bihqo", rotations, q_points) + translation[:, :, None, None, :]
key_points_global = torch.einsum("biop,bihqp->bihqo", rotations, k_points) + translation[:, :, None, None, :]
value_points_global = torch.einsum("biop,bihqp->bihqo", rotations, v_points) + translation[:, :, None, None, :]
bias = self.linear_bias(pair_representation)
attention_logits = torch.matmul(
q.permute(0, 2, 1, 3),
k.permute(0, 2, 3, 1),
)
attention_logits *= math.sqrt(1.0 / (3.0 * self.head_dim))
attention_logits += math.sqrt(1.0 / 3.0) * bias.permute(0, 3, 1, 2)
point_attention = query_points_global[:, :, None, :, :, :] - key_points_global[:, None, :, :, :, :]
point_attention = torch.sum(point_attention ** 2, dim=-1)
head_weights = torch.nn.functional.softplus(self.head_weights).view(1, 1, 1, self.num_heads, 1)
head_weights = head_weights * math.sqrt(1.0 / (3.0 * (self.n_query_points * 9.0 / 2.0)))
point_attention = torch.sum(point_attention * head_weights, dim=-1) * (-0.5)
attention_logits += point_attention.permute(0, 3, 1, 2)
if seq_mask is not None:
square_mask = seq_mask.unsqueeze(-1) * seq_mask.unsqueeze(-2)
attention_logits = attention_logits + self.inf * (square_mask[:, None, :, :] - 1.0)
attention = torch.nn.functional.softmax(attention_logits, dim=-1)
return self._assemble_output_features(
attention=attention,
value_scalar=v,
value_points_global=value_points_global,
pair_representation=pair_representation,
rotations=rotations,
translation=translation,
)
def forward(self, single_representation: torch.Tensor, pair_representation: torch.Tensor,
rotations: torch.Tensor, translation: torch.Tensor,
seq_mask: Optional[torch.Tensor] = None):
# single_rep shape: (batch, N_res, c_s)
# pair_rep shape: (batch, N_res, N_res, c_z)
# rotations shape: (batch, N_res, 3, 3)
# translations shape: (batch, N_res, 3)
# seq_mask shape: (batch, N_res) — 1 for valid, 0 for padding
assert rotations.shape[-2:] == (3, 3), \
f"rotations must end with (3, 3), got {rotations.shape}"
assert translation.shape[-1] == 3, \
f"translation must end with (3,), got {translation.shape}"
output_features = self._forward_output_features(
single_representation,
pair_representation,
rotations,
translation,
seq_mask=seq_mask,
)
output = self.linear_output(output_features)
# Zero out padded query positions
if seq_mask is not None:
output = output * seq_mask[:, :, None]
return outputThree things to look for as you read. First, the linear projections fall into three groups: scalar queries/keys/values (linear_q, linear_kv, etc.), 3D query/key/value points (linear_q_points, linear_kv_points), and the gate. The scalar projections produce tensors of shape ; the point projections produce , where the extra dimension is 3D space. Second, _project_points takes the point tensors and applies each residue’s frame to them. This is the operation in the equation. Third, the score is a sum of the three terms: Q·K dot product, pair bias projection, and squared-distance sum across points, with a learnable per-head weight controlling the point contribution.
The output is more elaborate than in standard attention. IPA returns (1) a weighted sum of scalar values, which is the regular attention output; (2) a weighted sum of value points, giving a point-cloud-like output with one 3D vector per head; and (3) scalar features derived from the attention-weighted norms of those value points. These get concatenated and projected back into the single representation . In other words, IPA returns scalar information and geometric information at the same time.
IPA is genuinely new. There were attention variants that operated on graphs or respected symmetries before AlphaFold2, but IPA’s particular trick — attention scores as a learnable mixture of scalar, pair-bias, and 3D-point-distance terms, with the point-distance term made invariant by local frames — was one of AF2’s major architectural contributions. You can see its influence in many later structure-prediction systems and reimplementations. When you see geometry-aware attention in this literature, IPA is one of the ancestors.
10c. Backbone update
IPA and its transition MLP produce an updated single representation for each residue. The backbone update is the step that turns this into an update on the frame . The mechanics are simple: project to six scalars, interpret the first three as the imaginary part of an unnormalized quaternion, interpret the last three as a translation, and compose the resulting rigid transformation into .
class BackboneUpdate(torch.nn.Module):
"""Backbone frame update (Algorithm 23, supplement 1.8.3).
Projects ``s_i`` to six scalars per residue: three for the local
axis-angle rotation update (interpreted as a unit quaternion
``(1, b, c, d)`` after normalising, line 3-4) and three for the
translation update (in the residue's own frame, line 5). The
current frame ``T_i`` is then updated as ``T_i ← T_i ∘ (R, t)``.
The output projection is ``final``-initialised (zero) so each IPA
iteration starts as a no-op — training discovers the updates.
"""
def __init__(self, config):
super().__init__()
self.linear = torch.nn.Linear(in_features=config.c_s, out_features=6)
_init_linear(self.linear, init="final")
def forward(self, single_representation: torch.Tensor):
# output shape; (batch, N_res, 6)
vals = self.linear(single_representation)
# Rotation quaternions
b = vals[:, :, 0]
c = vals[:, :, 1]
d = vals[:, :, 2]
a = torch.ones_like(b)
norm = torch.sqrt(1 + b**2 + c**2 + d**2)
a = a / norm
b = b / norm
c = c / norm
d = d / norm
# Construct pairwise multiplications for rotation matrix
aa = a*a
bb = b*b
cc = c*c
dd = d*d
ab = a*b
ac = a*c
ad = a*d
bc = b*c
bd = b*d
cd = c*d
# Construct rotation matrix entries
r11 = aa + bb - cc - dd
r12 = 2*bc - 2*ad
r13 = 2*bd + 2*ac
r21 = 2*bc + 2*ad
r22 = aa - bb + cc - dd
r23 = 2*cd - 2*ab
r31 = 2*bd - 2*ac
r32 = 2*cd + 2*ab
r33 = aa - bb - cc + dd
# Output shape: (batch, N_res, 3, 3)
R = torch.stack([r11, r12, r13, r21, r22, r23, r31, r32, r33], dim=-1).reshape((single_representation.shape[0], single_representation.shape[1], 3, 3))
t = vals[:, :, 3:]
return R, tThe quaternion construction is the clever bit. Rather than projecting to four scalars , AlphaFold2 projects to three scalars and forms . Why? Because at initialization, the projection is near zero, so and , the identity quaternion. Hence, the update is near-identity at initialization. Without this trick, the model would begin by predicting arbitrary rotations and would have to learn its way back to stability. With it, the model starts from “no change” and learns corrections.
Read that carefully — the backbone update is additive in the space of rotations, not in the space of quaternion components. The composition multiplies rotations (because isn’t a vector space) and adds translations. The QuaternionFrame widget from §10a lets you see concretely what small perturbations in do to the rotation matrix — set to small values and watch the rotation stay close to identity.
10d. All-atom coordinates
A per-residue backbone frame is not enough to call a protein structure predicted. We need all the atoms: backbone atoms (N, Cα, C, O) plus whatever side-chain atoms the residue type has. For the 20 canonical amino acids, this is represented as at most 14 atoms per residue, the “atom14” representation used throughout the codebase.
So how do we get from backbone frames to all atoms? AlphaFold2 reduces the remaining degrees of freedom to seven torsion angles per residue: the three backbone dihedrals (, , ) plus up to four side-chain angles. Given the residue type and these torsion angles, the full all-atom structure is determined up to fixed chemical geometry. Bond lengths and bond angles come from literature values in residue_constants.py; the torsions are what the model has to predict.
AlphaFold2 predicts the seven torsions per residue with a small head attached to the Structure Module output, then composes a chain of small rigid-body transformations to produce the atom coordinates:
def compute_all_atom_coordinates(
translations: torch.Tensor, # (batch, N_res, 3)
rotations: torch.Tensor, # (batch, N_res, 3, 3)
torsion_angles: torch.Tensor, # (batch, N_res, 7, 2) — [ω, φ, ψ, χ1, χ2, χ3, χ4]
aatype: torch.Tensor, # (batch, N_res) — integer residue type indices
default_frames: torch.Tensor, # (21, 8, 4, 4) — registered buffer
lit_positions: torch.Tensor, # (21, 14, 3) — registered buffer
atom_frame_idx_table: torch.Tensor, # (21, 14) — registered buffer
atom_mask_table: torch.Tensor, # (21, 14) — registered buffer
):
"""Assemble per-residue atom14 coordinates (Algorithm 24).
Given the backbone frame ``(R_i, t_i)`` and seven torsion angles
``α_i``, this rolls out the eight per-residue rigid-group frames
(backbone + ω + φ + ψ + χ1–χ4) and places each atom by looking up
which group it belongs to and its literature position within that
group:
* Lines 1-10: build the 8 frames parametrically — every sidechain
frame composes ``T_i ∘ T^lit_{r,f→bb} ∘ makeRotX(α_f)`` so the
torsion angles rotate about the local x-axis of each group
(:func:`make_rot_x`). χ2-χ4 chain off χ1 instead of the backbone.
Factored out into :func:`rigid_group_frames_from_torsions` so the
data pipeline can build ground-truth sidechain frames the same
way (see :func:`~minalphafold.data.build_supervision`).
* Lines 11-14: for each atom slot, look up its group index and
literature position, apply that group's frame.
Inputs are in the Structure Module's nm units; ``lit_positions``
and ``default_frames`` are already pre-scaled in
:class:`StructureModule.__init__`. Returns a dict with the 8
per-group frames (``frames_R``, ``frames_t``), the atom14 atom
positions ``atom_pos``, and per-atom validity ``atom_mask``.
"""
dtype = translations.dtype
device = translations.device
# Steps 1-4: build the 8 rigid-group frames (Algorithm 24 lines 1-10).
all_frames_R, all_frames_t = rigid_group_frames_from_torsions(
translations, rotations, torsion_angles, aatype, default_frames,
)
# --- Step 5: Place atoms using their frame assignments ---
lit_pos = lit_positions.to(device=device, dtype=dtype)[aatype] # (batch, N_res, 14, 3)
atom_frame_idx = atom_frame_idx_table.to(device=aatype.device)[aatype] # (batch, N_res, 14)
mask = atom_mask_table.to(device=device, dtype=dtype)[aatype] # (batch, N_res, 14)
# Gather the correct frame for each atom
idx_R = atom_frame_idx[:, :, :, None, None].expand(-1, -1, -1, 3, 3)
atom_R = torch.gather(all_frames_R, 2, idx_R) # (batch, N_res, 14, 3, 3)
idx_t = atom_frame_idx[:, :, :, None].expand(-1, -1, -1, 3)
atom_t = torch.gather(all_frames_t, 2, idx_t) # (batch, N_res, 14, 3)
# x_global = R_frame @ x_lit + t_frame
atom_coords = torch.einsum('bnaij, bnaj -> bnai', atom_R, lit_pos) + atom_t
return all_frames_R, all_frames_t, atom_coords, maskFor each residue, eight rigid groups are assembled:
- Backbone (N, Cα, C, O): sits in the residue’s backbone frame directly.
- Group for : positioned by rotating around a literature-defined axis by the predicted angle, with literature-defined atoms attached.
- Group for : positioned relative to group by another torsion rotation.
- Groups for , : same pattern.
- Three more groups for secondary geometry (e.g., ring planes).
def rigid_group_frames_from_torsions(
translations: torch.Tensor, # (batch, N_res, 3)
rotations: torch.Tensor, # (batch, N_res, 3, 3)
torsion_angles: torch.Tensor, # (batch, N_res, 7, 2) — [ω, φ, ψ, χ1, χ2, χ3, χ4]
aatype: torch.Tensor, # (batch, N_res)
default_frames: torch.Tensor, # (21, 8, 4, 4)
) -> tuple[torch.Tensor, torch.Tensor]:
"""Build the 8 per-residue rigid-group frames parametrically (Algorithm 24).
This is the frame-construction half of ``compute_all_atom_coordinates``,
factored out so ``data.build_supervision`` can build **ground-truth**
rigid-group frames the same way the Structure Module builds its
**predicted** frames. If the two paths diverge (e.g. GT frames built
via Gram-Schmidt on real atoms), the sidechain FAPE loss acquires a
non-zero floor equal to the bond-length-idealisation mismatch between
literature geometry and real atoms — even when the prediction exactly
matches the ground truth. Building both paths parametrically makes that
floor vanish (down to atom-level idealisation only, which is tiny).
Returns ``(all_frames_R, all_frames_t)`` of shapes ``(batch, N_res, 8,
3, 3)`` and ``(batch, N_res, 8, 3)``. Group order matches Algorithm 24:
``[backbone, ω, φ, ψ, χ1, χ2, χ3, χ4]``.
"""
dtype = translations.dtype
device = translations.device
# Normalize torsion angles to unit vectors (Algorithm 24 line 1).
torsion_angles = torsion_angles / (torch.norm(torsion_angles, dim=-1, keepdim=True) + 1e-8)
# Per-residue-type literature transforms (Algorithm 24 line 2): T^lit_{r,*→bb}.
lit_all = default_frames.to(device=device, dtype=dtype)[aatype] # (batch, N_res, 8, 4, 4)
lit_R = lit_all[..., :3, :3] # (batch, N_res, 8, 3, 3)
lit_t = lit_all[..., :3, 3] # (batch, N_res, 8, 3)
# Torsion rotations via makeRotX (Algorithm 25).
torsion_R, torsion_t = make_rot_x(torsion_angles) # (batch, N_res, 7, 3, 3), (batch, N_res, 7, 3)
frames_R = [rotations] # Frame 0: backbone
frames_t = [translations]
# Frames 1-4: Algorithm 24 lines 4-7 — ω, φ, ψ, χ1 each branch off the
# backbone frame via its own literature transform + torsion rotation.
for f in range(4):
mid_R, mid_t = compose_transforms(
lit_R[:, :, f + 1], lit_t[:, :, f + 1],
torsion_R[:, :, f], torsion_t[:, :, f],
)
frame_R, frame_t = compose_transforms(rotations, translations, mid_R, mid_t)
frames_R.append(frame_R)
frames_t.append(frame_t)
# Frames 5-7: Algorithm 24 lines 8-10 — χ2 chains off χ1, χ3 off χ2, χ4 off χ3.
for f in range(3):
prev_R = frames_R[f + 4]
prev_t = frames_t[f + 4]
mid_R, mid_t = compose_transforms(
lit_R[:, :, f + 5], lit_t[:, :, f + 5],
torsion_R[:, :, f + 4], torsion_t[:, :, f + 4],
)
frame_R, frame_t = compose_transforms(prev_R, prev_t, mid_R, mid_t)
frames_R.append(frame_R)
frames_t.append(frame_t)
all_frames_R = torch.stack(frames_R, dim=2) # (batch, N_res, 8, 3, 3)
all_frames_t = torch.stack(frames_t, dim=2) # (batch, N_res, 8, 3)
return all_frames_R, all_frames_tThe make_rot_x helper does the geometric work. It rotates around the local -axis by a learned angle encoded as sine and cosine, rather than as a raw angle, so the model does not have to deal with wrap-around at . Each rigid-group frame is computed as : compose the parent group’s frame with the literature default transformation and then with the torsion-angle rotation.
Once the eight group frames are known, the actual atom positions are looked up from residue_constants.py’s restype_atom14_rigid_group_positions table. This table says, for each amino acid, “atom at index of atom14 sits at local coordinates in rigid group .” Transform by the group frame and you have the atom’s global position.
The torsion head and atom-coordinate composition are surprisingly simple given what they accomplish. The whole thing — 8 rigid groups, 7 torsion angles per residue, and a lookup table of atom positions — fits in a few hundred lines. The reason this works is the separation of concerns. The Structure Module does not need to learn bond lengths or bond angles; chemistry supplies those. It only has to learn where to place each residue’s backbone frame and which torsion angles to emit. The residue-constant tables do the rest.
10e. The full structure module loop
With IPA, the backbone update, and the all-atom construction in place, the Structure Module itself is a short loop: eight iterations, weights shared across iterations, with each iteration refining the backbone frames and side-chain torsions.
class StructureModule(torch.nn.Module):
"""Structure Module — iterative IPA + frame update (Algorithm 20).
Given the Evoformer's ``s_i`` and ``z_ij``:
1. Normalise inputs and project ``s_i`` to the module's internal
``c`` channel dim (line 1-2).
2. Initialise the per-residue rigid frame ``T_i`` to the identity
("black-hole initialisation", line 3).
3. Run ``config.structure_module_layers`` IPA iterations (default
8): :class:`InvariantPointAttention` updates ``s_i`` with
geometric context; a transition MLP mixes channels; a
:class:`BackboneUpdate` applies a local rotation + translation
to ``T_i`` (supplement 1.8.3).
4. After every iteration, :class:`MultiRigidSidechain` predicts
seven torsion angles from ``s_i`` and :func:`compute_all_atom_coordinates`
rolls them + ``T_i`` out to atom14 coordinates (Algorithm 24).
Units: internal nm (literature bond lengths divided by
``position_scale``, output translations multiplied back to Å).
The frame rotation is detached between iterations (line 13,
"stop_gradient" on rotation only) — translations keep gradients so
the auxiliary FAPE on Cα at every layer has signal through to the
Evoformer.
"""
default_frames: torch.Tensor
lit_positions: torch.Tensor
atom_frame_idx_table: torch.Tensor
atom_mask_table: torch.Tensor
def __init__(self, config):
super().__init__()
self.c = config.structure_module_c
self.num_layers = config.structure_module_layers
self.position_scale = float(getattr(config, "position_scale", 10.0))
# Layer Norms
self.layer_norm_single_rep_1 = torch.nn.LayerNorm(config.c_s)
self.layer_norm_single_rep_2 = torch.nn.LayerNorm(config.c_s)
self.layer_norm_single_rep_3 = torch.nn.LayerNorm(config.c_s)
self.layer_norm_pair_rep = torch.nn.LayerNorm(config.c_z)
# Dropouts (rates from config)
self.dropout_1 = torch.nn.Dropout(p=config.structure_module_dropout_ipa)
self.dropout_2 = torch.nn.Dropout(p=config.structure_module_dropout_transition)
# Register residue constant tensors as buffers (avoid device bugs, improve speed)
self.register_buffer('default_frames',
torch.tensor(restype_rigid_group_default_frame)) # (21, 8, 4, 4)
self.register_buffer('lit_positions',
torch.tensor(restype_atom14_rigid_group_positions)) # (21, 14, 3)
self.register_buffer('atom_frame_idx_table',
torch.tensor(restype_atom14_to_rigid_group)) # (21, 14)
self.register_buffer('atom_mask_table',
torch.tensor(restype_atom14_mask)) # (21, 14)
# Linear layers
self.single_rep_proj = torch.nn.Linear(in_features=config.c_s, out_features=config.c_s)
self.transition_linear_1 = torch.nn.Linear(in_features=config.c_s, out_features=config.c_s)
self.transition_linear_2 = torch.nn.Linear(in_features=config.c_s, out_features=config.c_s)
self.transition_linear_3 = torch.nn.Linear(in_features=config.c_s, out_features=config.c_s)
_init_linear(self.single_rep_proj, init="default")
_init_linear(self.transition_linear_1, init="relu")
_init_linear(self.transition_linear_2, init="relu")
_init_linear(self.transition_linear_3, init="final")
# Core blocks
self.IPA = InvariantPointAttention(config)
self.backbone_update = BackboneUpdate(config)
self.sidechain_module = MultiRigidSidechain(config)
self.relu = torch.nn.ReLU()
# AF2 keeps structure-module translations in internal units and rescales
# them by position_scale when materializing coordinates and losses.
internal_scale = 1.0 / self.position_scale
self.default_frames[..., :3, 3] *= internal_scale
self.lit_positions *= internal_scale
def forward(self, single_representation: torch.Tensor, pair_representation: torch.Tensor,
aatype: torch.Tensor, seq_mask: Optional[torch.Tensor] = None,
detach_rotations: bool = True):
# seq_mask: (batch, N_res) — 1 for valid residues, 0 for padding
# detach_rotations: if True (default, AF2 standard), apply stopgrad to
# the rotation component of T_i between iterations (Algorithm 20
# lines 19-21). The detach is placed at the *end* of each non-final
# iteration so that the next iteration's IPA sees T_i with no
# rotation-gradient path, preventing lever effects through the
# chained composition of frames. Set to False to allow full gradient
# flow (useful for memorization/debugging).
assert single_representation.ndim == 3, \
f"single_representation must be (batch, N_res, c_s), got {single_representation.shape}"
assert pair_representation.ndim == 4, \
f"pair_representation must be (batch, N_res, N_res, c_z), got {pair_representation.shape}"
assert aatype.ndim == 2, \
f"aatype must be (batch, N_res), got {aatype.shape}"
assert single_representation.shape[1] == pair_representation.shape[1] == pair_representation.shape[2] == aatype.shape[1], \
f"N_res mismatch: single={single_representation.shape[1]}, pair={pair_representation.shape[1:3]}, aatype={aatype.shape[1]}"
single_representation = self.layer_norm_single_rep_1(single_representation)
initial_single_representation = single_representation
pair_representation = self.layer_norm_pair_rep(pair_representation)
s = self.single_rep_proj(single_representation)
rotations = torch.eye(3, device=s.device, dtype=s.dtype).view(1, 1, 3, 3).expand(s.shape[0], s.shape[1], 3, 3)
translations = torch.zeros(s.shape[0], s.shape[1], 3, device=s.device, dtype=s.dtype)
# Collect intermediates for auxiliary losses
all_rotations = []
all_translations = []
all_torsion_angles = []
all_torsion_angles_unnormalized = []
sidechain_outputs = None
for l in range(self.num_layers):
# Algorithm 20, line 6-7: s += IPA(s); s = LN(Dropout(s))
s = s + self.IPA(s, pair_representation, rotations, translations, seq_mask)
s = self.layer_norm_single_rep_3(self.dropout_1(s))
# Algorithm 20, line 8-9: s += Transition(s); s = LN(Dropout(s))
s = s + self.transition_linear_3(self.relu(self.transition_linear_2(self.relu(self.transition_linear_1(s)))))
s = self.layer_norm_single_rep_2(self.dropout_2(s))
# Algorithm 20, line 10: T_i ← T_i ∘ BackboneUpdate(s_i)
new_rotations, new_translations = self.backbone_update(s)
translations = torch.einsum('bsij, bsj -> bsi', rotations, new_translations) + translations
rotations = torch.einsum('bsij, bsjk -> bsik', rotations, new_rotations)
# Algorithm 20, lines 11-18: side-chain torsions and auxiliary losses use
# the updated (un-detached) T_i so gradients reach this iteration's s.
sidechain_outputs = self.sidechain_module(
s,
initial_single_representation,
rotations,
translations,
aatype,
self.default_frames,
self.lit_positions,
self.atom_frame_idx_table,
self.atom_mask_table,
)
all_rotations.append(rotations)
all_translations.append(translations)
all_torsion_angles.append(sidechain_outputs["angles_sin_cos"])
all_torsion_angles_unnormalized.append(sidechain_outputs["unnormalized_angles_sin_cos"])
# Algorithm 20, lines 19-21: stopgrad on rotations between iterations
# (but not after the final one). Detaching *after* the backbone update
# means iteration l+1 receives the rotation without a gradient path,
# exactly as the supplement prescribes.
if detach_rotations and l < self.num_layers - 1:
rotations = rotations.detach()
all_rotations = torch.stack(all_rotations)
all_translations = torch.stack(all_translations)
all_torsion_angles = torch.stack(all_torsion_angles)
all_torsion_angles_unnormalized = torch.stack(all_torsion_angles_unnormalized)
if sidechain_outputs is None:
raise RuntimeError("StructureModule produced no sidechain outputs.")
all_frames_R = sidechain_outputs["frames_R"]
all_frames_t = sidechain_outputs["frames_t"]
atom_coords = sidechain_outputs["atom_pos"]
mask = sidechain_outputs["atom_mask"]
# Convert internal structure-module units back to angstroms.
# Rotations are unitless — no conversion needed
predictions = {
# Per-layer backbone frames for auxiliary FAPE loss
"traj_rotations": all_rotations, # (num_layers, batch, N_res, 3, 3)
"traj_translations": all_translations * self.position_scale,
# Per-layer torsion angles for torsion angle loss
"traj_torsion_angles": all_torsion_angles, # (num_layers, batch, N_res, 7, 2)
"traj_torsion_angles_unnormalized": all_torsion_angles_unnormalized,
# Final backbone frames
"final_rotations": rotations, # (batch, N_res, 3, 3)
"final_translations": translations * self.position_scale,
# Final all-atom outputs (8 rigid-group frames including backbone frame 0)
"all_frames_R": all_frames_R, # (batch, N_res, 8, 3, 3)
"all_frames_t": all_frames_t * self.position_scale,
"atom14_coords": atom_coords * self.position_scale,
"atom14_mask": mask, # (batch, N_res, 14)
# Final single representation (for distogram, pLDDT, etc.)
"single": s, # (batch, N_res, c_s)
}
return predictionsTwo details are worth noticing. First, every iteration applies IPA, transition, and backbone update, and all eight iterations see the same pair representation . The pair representation is computed once after the Evoformer and held fixed for the whole structure loop. What changes iteration by iteration is the set of frames, which feed back into IPA’s point-distance term and shift the attention pattern as the structure settles. Second, every iteration produces a full prediction: backbone frames and torsion-derived coordinates. Per-iteration auxiliary FAPE losses are applied during training, so the model is pressured to make every iteration structurally meaningful, not just the last one.
This is different from a vanilla encoder, where only the final layer is typically supervised. Applying FAPE at every iteration has two effects. First, it prevents early iterations from becoming arbitrary scratch space used only by the final iteration. Second, it lets the model act as a progressive refiner at inference time, producing a meaningful structure at every step. Combined with weight sharing, the Structure Module is best viewed as an unrolled iterative refinement process.
After the loop finishes, the Structure Module emits the final frames , the final atom14 coordinates, and the per-iteration predictions used for auxiliary losses. These, plus the final single and pair representations, feed into the auxiliary heads (§11). Critically, the final pseudo-Cβ positions also feed back into the recycling loop (§9) for the next cycle.
11. Auxiliary heads
The Structure Module produces the thing we ultimately care about: atom coordinates. But AlphaFold2 also emits a handful of auxiliary predictions from small heads attached to the final representations. Most of these exist to provide additional supervision during training, while some also give users useful information at inference.
The heads all live in a single short file:
| Head | Input | Output | Purpose |
|---|---|---|---|
DistogramHead | pair rep | 64-bin distribution over – distances | Dense structural supervision; predicts a probability distribution over pairwise distances. |
PLDDTHead | single rep | 50-bin distribution over per-residue lDDT | Model’s own confidence estimate; what gets output as the pLDDT score in predicted PDBs. |
MaskedMSAHead | full MSA rep | 23-way classification per masked token | BERT-style reconstruction signal on randomly masked MSA residues. Training-time only. |
ExperimentallyResolvedHead | single rep | per-atom binary mask | Predicts which atoms were experimentally resolved in the X-ray crystal. Helps the model down-weight loosely-defined regions in training. |
TMScoreHead | pair rep | 64-bin PAE matrix | Predicted Aligned Error — pairwise expected error if the predicted structure were aligned at residue . This is the output that justifies “this part of the prediction is trustworthy.” |
class DistogramHead(torch.nn.Module):
"""Distogram head (supplement 1.9.8).
Projects the pair representation ``z_ij`` to ``n_dist_bins`` bin
logits and symmetrises across (i, j). The loss target is one-hot
binned Cβ-Cβ (or Cα for glycine) distance between ground-truth
residue pairs, cross-entropy averaged (eq 41).
"""
def __init__(self, config):
super().__init__()
self.linear = torch.nn.Linear(config.c_z, config.n_dist_bins)
# Supplement 1.11.4: zero-init residue distance prediction logits
_zero_init_linear(self.linear)
def forward(self, pair_representation: torch.Tensor):
# pair_representation: (batch, N_res, N_res, c_z)
logits = self.linear(pair_representation) # (batch, N_res, N_res, n_dist_bins)
# Distograms of an unordered pair are symmetric by definition; average
# the (i, j) and (j, i) logits so predictions match that invariance.
logits = (logits + logits.transpose(1, 2)) / 2
return logitsEvery head is architecturally simple: a LayerNorm, a linear projection (sometimes two), and a softmax or sigmoid. The interesting part is not the head architecture but the target each head is trained against, which brings us to the losses in §12. The key architectural point is that the distogram and PAE heads both read from , not from the atom coordinates directly. The pair representation has to simultaneously (a) drive the Structure Module’s IPA through pair bias and (b) carry enough geometric information that pairwise distances and aligned errors can be read from it. This dual responsibility is part of why the pair stack is so expensive in the Evoformer. is doing a lot of work.
class PLDDTHead(torch.nn.Module):
"""Per-residue confidence head (Algorithm 29 / supplement 1.9.6).
Takes the post-Structure-Module single representation ``s_i``, normal-
ises it, passes it through a two-layer ReLU MLP, and projects to
``n_plddt_bins`` (default 50). The scalar pLDDT for residue i is
``sum_k p_i^k · v_k`` where ``v_k = k + 0.5`` scaled into [1, 99] —
that bin-centre transform lives in ``pdbio`` (B-factor write path) and
``losses.PLDDTLoss`` (LDDT-Cα supervision).
"""
def __init__(self, config):
super().__init__()
self.net = torch.nn.Sequential(
torch.nn.LayerNorm(config.c_s),
torch.nn.Linear(config.c_s, config.plddt_hidden_dim),
torch.nn.ReLU(),
torch.nn.Linear(config.plddt_hidden_dim, config.plddt_hidden_dim),
torch.nn.ReLU(),
torch.nn.Linear(config.plddt_hidden_dim, config.n_plddt_bins),
)
# Supplement 1.11.4: zero-init model confidence prediction logits
final_linear: torch.nn.Linear = self.net[-1] # type: ignore[assignment]
_zero_init_linear(final_linear)
def forward(self, single_representation: torch.Tensor):
# single_representation: (batch, N_res, c_s)
return self.net(single_representation) # (batch, N_res, n_plddt_bins)The pLDDT head is the one casual users interact with most. The coloring you see on predicted structures in the AlphaFold database — blue for “very high confidence,” green/yellow/orange/red descending — is per-residue pLDDT, with the bin centers averaged into a single scalar. Under the hood, it’s a 50-way classifier trained against lDDT- scores computed against the ground truth structure (lDDT is a local, alignment-free similarity metric that roughly measures the fraction of residue pairs whose predicted distance is close to the true distance, with a tolerance that shrinks as the distance shrinks). In essence, the model is learning to predict its own error.
12. Losses
The loss is where AlphaFold2 spends a surprising amount of its design budget. Structure prediction has a property that makes the loss non-trivial: the output lives in 3D space, where a naive distance between predicted and true coordinates depends on the arbitrary global orientation of both structures. If you predict a structure that is correct but globally rotated 90°, coordinate MSE will punish you heavily, even though the prediction is chemically and biologically the same.
AlphaFold2’s solution is Frame-Aligned Point Error, or FAPE. It is the central loss in the paper and the centerpiece of this section. After FAPE, we will cover the remaining terms more quickly: torsion loss, pLDDT loss, distogram loss, structural violations, and the combined AlphaFoldLoss that weights them together.
12.1 FAPE
The idea of FAPE is simple once you have internalized rigid frames from §10. Rather than comparing predicted and true atom positions in one global coordinate system, FAPE compares them in every residue’s local frame, one at a time, and averages. Concretely, for a predicted frame and predicted atom position , along with their ground-truth counterparts and , the per-pair FAPE error is:
Read the two terms from inside out. is “the position of atom as seen by residue ‘s local frame,” exactly the same local-frame operation we met in IPA (§10b). is the same thing in the ground truth structure: where residue should have seen atom . The error is the Euclidean distance between those two local-frame vectors.
This has two crucial properties. First, it is SE(3) invariant by construction. Apply any global rigid motion to the predicted structure. Frames transform as and atoms as . The local-frame position becomes . Same answer. The loss does not penalize global rotations or translations. Second, it is per-residue: the error compares residue ‘s predicted local view to residue ‘s true local view. Every residue gets its own local judgment of the structure.
The invariance is easier to feel than to derive. The widget below puts two chains side by side: ground truth in gray, prediction in pink. One slider rotates the whole prediction globally; the other pushes a single residue away from its true position. Coordinate MSE and FAPE update in real time. As you rotate the prediction, coordinate MSE rises because every residue has moved in the global frame, while FAPE stays fixed because every local-frame relationship is preserved. Push the single residue, and both losses rise. This is the whole argument for FAPE in one interaction: insensitive to arbitrary global orientation, sensitive to actual structural mistakes.
def frame_aligned_point_error(
predicted_rotations: torch.Tensor,
predicted_translations: torch.Tensor,
true_rotations: torch.Tensor,
true_translations: torch.Tensor,
predicted_positions: torch.Tensor,
true_positions: torch.Tensor,
frames_mask: torch.Tensor,
positions_mask: torch.Tensor,
*,
length_scale: float,
pair_mask: Optional[torch.Tensor] = None,
l1_clamp_distance: Optional[float] = None,
eps: float,
) -> torch.Tensor:
"""Frame aligned point error (Algorithm 28, supplement 1.9.2).
Computes `L_FAPE = (1/Z) mean_{i,j}(min(d_clamp, d_ij))` where
`d_ij = sqrt(||T_i^{-1} ∘ x_j - T_i^{true -1} ∘ x_j^{true}||^2 + eps)`.
ε is a caller-supplied smoothing constant. Algorithm 20 uses ε = 10⁻¹²
for the per-layer auxiliary FAPE on Cα atoms (line 17) and ε = 10⁻⁴ for
the final all-atom FAPE (line 28); the paper notes the exact value does
not matter as long as it is small enough. `length_scale` is Z = 10 Å
(supplement 1.9.2). `l1_clamp_distance` is d_clamp = 10 Å when clamping
is requested (supplement 1.11.5).
Masks extend the paper to accommodate padded/variable-length batches:
`frames_mask` and `positions_mask` mark valid frames i and atoms j; the
optional `pair_mask` also masks specific (i, j) pairs. The denominator
is the number of valid (i, j) pairs — equal to `N_res^2` when nothing
is masked, matching the supplement's `mean_{i,j}`.
"""
# Algorithm 28 lines 1-2: x_ij = T_i^{-1} ∘ x_j, with (R, t)^{-1} = (R^T, -R^T t).
predicted_rotations_inv = predicted_rotations.transpose(-1, -2)
predicted_translations_inv = -torch.einsum(
"...ij,...j->...i",
predicted_rotations_inv,
predicted_translations,
)
true_rotations_inv = true_rotations.transpose(-1, -2)
true_translations_inv = -torch.einsum(
"...ij,...j->...i",
true_rotations_inv,
true_translations,
)
local_predicted_positions = (
torch.einsum("...fij,...aj->...fai", predicted_rotations_inv, predicted_positions)
+ predicted_translations_inv[..., :, None, :]
)
local_true_positions = (
torch.einsum("...fij,...aj->...fai", true_rotations_inv, true_positions)
+ true_translations_inv[..., :, None, :]
)
# Algorithm 28 line 3: d_ij = sqrt(||Δx||^2 + ε).
error_distance = torch.sqrt(
torch.sum((local_predicted_positions - local_true_positions) ** 2, dim=-1) + eps
)
# Algorithm 28 line 4: min(d_clamp, d_ij) is equivalent to clamp(max=d_clamp)
# since d_ij ≥ 0 by construction.
if l1_clamp_distance is not None:
error_distance = error_distance.clamp(max=l1_clamp_distance)
# mean_{i,j}(...) divided by length_scale Z. The mask zeros out (i, j)
# pairs where either the frame or atom is invalid (or the caller-provided
# pair_mask rejects the pair); the denominator counts just the surviving
# pairs, so un-masked inputs recover the paper's 1/N_res^2 normalisation.
mask = frames_mask[..., :, None] * positions_mask[..., None, :]
if pair_mask is not None:
mask = mask * pair_mask
numerator = (error_distance * mask).sum(dim=(-1, -2))
denominator = mask.sum(dim=(-1, -2)).clamp(min=1.0)
return numerator / (denominator * length_scale)Two implementation details matter here.
The distance clamp. The raw error can be arbitrarily large. If the model gets a structure very wrong, some local-frame distances can blow up to hundreds of angstroms. FAPE clamps at d_clamp, default 10 Å. This is not cosmetic. Without clamping, the gradient from a small number of badly misplaced atoms can dominate the whole update. With clamping, the model is first pressured to bring all atoms within the relevant neighborhood before it is rewarded for making already-close atoms even closer.
The normalization. FAPE divides the sum of clamped distances by d_clamp × mask.sum(), so the loss is dimensionless and in [0, 1]. This is a small thing, but it matters for how weights in the combined loss are chosen — every term stays on the same scale.
12.2 Per-iteration backbone FAPE
The Structure Module’s 8 iterations each produce a set of backbone frames (§10e). During training, FAPE is evaluated on every iteration’s output — not just the last one — and the per-iteration losses are averaged into a single “backbone trajectory” loss.
class BackboneTrajectoryLoss(torch.nn.Module):
"""Per-iteration backbone FAPE averaged over layers (L_aux^{FAPE}).
Algorithm 20 emits backbone frames at every iteration l ∈ [1, N_layer];
line 17 computes a Cα-only FAPE against ground truth on each iteration
and line 23 averages them to yield the FAPE component of L_aux.
`use_clamped_fape ∈ [0, 1]` (supplement 1.11.5): weight of the clamped
FAPE in a soft mix with the unclamped version. `None` ≡ 1.0 (fully
clamped). AlphaFold samples 10% of mini-batches to be fully unclamped,
so the expected loss per batch has ≈0.9 weight on the clamped form;
passing `use_clamped_fape=0.9` reproduces that expectation directly.
"""
def __init__(self):
super().__init__()
self.fape_loss = BackboneFAPE()
def forward(
self,
structure_model_prediction: dict,
true_rotations: torch.Tensor, # (b, N_res, 3, 3)
true_translations: torch.Tensor, # (b, N_res, 3)
backbone_mask: Optional[torch.Tensor] = None,
seq_mask: Optional[torch.Tensor] = None,
use_clamped_fape: Optional[torch.Tensor] = None,
):
traj_R = structure_model_prediction["traj_rotations"] # (L, b, N_res, 3, 3)
traj_t = structure_model_prediction["traj_translations"] # (L, b, N_res, 3)
num_layers = traj_R.shape[0]
total_loss = torch.zeros(traj_R.shape[1], device=traj_R.device, dtype=traj_R.dtype)
if backbone_mask is None:
valid_mask = seq_mask
elif seq_mask is None:
valid_mask = backbone_mask
else:
valid_mask = backbone_mask * seq_mask
for l in range(num_layers):
clamped_fape = self.fape_loss(
traj_R[l],
traj_t[l],
true_rotations,
true_translations,
frame_mask=valid_mask,
position_mask=valid_mask,
l1_clamp_distance=self.fape_loss.d_clamp_val,
)
if use_clamped_fape is None:
total_loss = total_loss + clamped_fape
else:
unclamped_fape = self.fape_loss(
traj_R[l],
traj_t[l],
true_rotations,
true_translations,
frame_mask=valid_mask,
position_mask=valid_mask,
l1_clamp_distance=None,
)
total_loss = total_loss + (
clamped_fape * use_clamped_fape + unclamped_fape * (1.0 - use_clamped_fape)
)
# Algorithm 20 line 23: L_aux = mean_l(L_aux^l). This returns just the
# FAPE component; the torsion component is added separately by the
# caller as TorsionAngleLoss.
return total_loss / num_layersThis is a meaningful design choice. It pressures every iteration to produce a structurally reasonable prediction, not just the final one. Without this, early iterations could emit arbitrary intermediate frames that are useful only to later iterations. With per-iteration FAPE, each iteration is trained to be an incremental refinement.
12.3 All-atom FAPE
The final piece is all-atom FAPE. Once the Structure Module has emitted its last set of backbone frames and the all-atom reconstruction (§10d) has placed every side-chain atom, FAPE is evaluated on the full atom14 tensor — every backbone and side-chain atom of every residue, as seen by every residue’s backbone frame.
class AllAtomFAPE(torch.nn.Module):
"""Final all-atom FAPE (Algorithm 20 line 28).
Scores the 8 per-residue rigid-group frames (3 backbone + 4 side-chain
torsion frames + ψ frame, see Table 2) against all 14 atom positions of
every residue after the symmetric-ground-truth renaming of Algorithm 26.
Per supplement 1.11.5, the side-chain FAPE is *always* clamped by
d_clamp = 10 Å (unlike the backbone FAPE, which is unclamped in 10% of
mini-batches). Uses ε = 10⁻⁴ per Algorithm 20 line 28.
"""
def __init__(self, d_clamp: float = 10.0, eps: float = 1e-4, Z: float = 10.0):
super().__init__()
self.eps = eps
self.d_clamp_val = d_clamp
self.Z = Z
def forward(self,
predicted_frames_R, # (b, N_res, 8, 3, 3)
predicted_frames_t, # (b, N_res, 8, 3)
predicted_atom_positions, # (b, N_res, 14, 3)
atom_mask, # (b, N_res, 14) — predicted atom existence
true_frames_R, # (b, N_res, 8, 3, 3)
true_frames_t, # (b, N_res, 8, 3)
true_atom_positions, # (b, N_res, 14, 3)
true_atom_mask: Optional[torch.Tensor] = None, # (b, N_res, 14)
seq_mask: Optional[torch.Tensor] = None, # (b, N_res)
frame_mask: Optional[torch.Tensor] = None, # (b, N_res, 8)
):
b, N_res, n_frames = predicted_frames_R.shape[:3]
n_atoms = predicted_atom_positions.shape[2]
# Flatten per-residue rigid groups and atoms into a single list of
# frames / atoms so FAPE scores every (frame, atom) pair, as required
# by Algorithm 20 line 28's `mean_{i,j}` over the full structure.
pred_R = predicted_frames_R.reshape(b, N_res * n_frames, 3, 3)
pred_t = predicted_frames_t.reshape(b, N_res * n_frames, 3)
true_R = true_frames_R.reshape(b, N_res * n_frames, 3, 3)
true_t = true_frames_t.reshape(b, N_res * n_frames, 3)
pred_pos = predicted_atom_positions.reshape(b, N_res * n_atoms, 3)
true_pos = true_atom_positions.reshape(b, N_res * n_atoms, 3)
flat_atom_mask = atom_mask.reshape(b, N_res * n_atoms)
if true_atom_mask is not None:
flat_atom_mask = flat_atom_mask * true_atom_mask.reshape(b, N_res * n_atoms)
group_mask = (
frame_mask.to(predicted_frames_R.dtype)
if frame_mask is not None
else predicted_frames_R.new_ones(b, N_res, n_frames)
)
# Fold the residue-level seq_mask into both the per-atom mask and
# per-frame mask so padded residues do not contribute.
if seq_mask is not None:
seq_atom_mask = seq_mask[:, :, None].expand(-1, -1, n_atoms).reshape(b, N_res * n_atoms)
flat_atom_mask = flat_atom_mask * seq_atom_mask
flat_frame_mask = (seq_mask[:, :, None] * group_mask).reshape(b, N_res * n_frames)
else:
flat_frame_mask = group_mask.reshape(b, N_res * n_frames)
return frame_aligned_point_error(
predicted_rotations=pred_R,
predicted_translations=pred_t,
true_rotations=true_R,
true_translations=true_t,
predicted_positions=pred_pos,
true_positions=true_pos,
frames_mask=flat_frame_mask,
positions_mask=flat_atom_mask,
length_scale=self.Z,
# Supplement 1.11.5: side-chain FAPE is always clamped.
l1_clamp_distance=self.d_clamp_val,
eps=self.eps,
)One subtlety is worth noting. The reference frames for this loss come from the true backbone atoms, not the predicted ones. And since some residues have symmetric side chains, the ground truth is “renamed” through Algorithm 26 of the supplement to match whichever chemically equivalent atom permutation the model predicted. That permutation search is handled upstream of this loss, so by the time FAPE runs, the predicted and true atom tensors are in the right correspondence.
12.4 Torsion angle loss
Side-chain torsions ( through ) and backbone torsions (, , ) need their own supervision. Torsion angles live on the circle , so the naive approach, L2 on the raw angle, has a discontinuity at . AlphaFold2 avoids this by predicting pairs and supervising those:
class TorsionAngleLoss(torch.nn.Module):
"""Side-chain and backbone torsion angle loss (Algorithm 27).
Scores all 7 torsions f ∈ {ω, φ, ψ, χ1, χ2, χ3, χ4} per the supplement.
Validity of each torsion is carried by `torsion_mask_true` (shape
[..., 7]): ω/φ are undefined for the first residue, χ1..χ4 existence
depends on residue type, and any torsion whose atoms are missing in the
ground truth is masked out. See `geometry.torsion_angles` for how the
mask is built from atom14 data.
The min-of-(true, alt_true) term in line 3 handles 180°-rotation symmetry
for ASP/GLU/PHE/TYR; for all other torsions alt_true == true, so the
minimum reduces to the plain L2 term.
Both `torsion_weight` and `angle_norm_weight` pre-apply the outer 0.5
factor that equation (7) multiplies L_aux by: they are (1.0, 0.02) from
Algorithm 27 times 0.5, i.e. (0.5, 0.01). Keeping the pre-multiplication
here lets `AlphaFoldLoss` add the returned value directly.
"""
def __init__(self):
super().__init__()
self.torsion_weight = 0.5
self.angle_norm_weight = 0.01
def forward(
self,
torsion_angles: torch.Tensor,
unnormalized_torsion_angles: torch.Tensor,
torsion_angles_true: torch.Tensor,
torsion_angles_true_alt: torch.Tensor,
torsion_mask_true: torch.Tensor,
seq_mask: Optional[torch.Tensor] = None,
):
# Prepend the per-layer trajectory dim if only the final iteration was
# passed (Algorithm 20 averages L_aux over layers; the normalization
# below sums across L so a single layer broadcasts as L=1).
if torsion_angles.ndim == 4:
torsion_angles = torsion_angles.unsqueeze(0)
unnormalized_torsion_angles = unnormalized_torsion_angles.unsqueeze(0)
true_angles = torsion_angles_true.unsqueeze(0)
true_alt = torsion_angles_true_alt.unsqueeze(0)
mask = torsion_mask_true.unsqueeze(0) # (1, b, N_res, 7)
if seq_mask is not None:
mask = mask * seq_mask.unsqueeze(0).unsqueeze(-1)
# Algorithm 27 line 3: L_torsion = mean_{i,f} min(||α̂ - α_true||², ||α̂ - α_alt||²).
true_dist_sq = torch.sum((true_angles - torsion_angles) ** 2, dim=-1)
alt_dist_sq = torch.sum((true_alt - torsion_angles) ** 2, dim=-1)
torsion_dist_sq = torch.minimum(true_dist_sq, alt_dist_sq)
torsion_normalizer = mask.sum(dim=(0, 2, 3)).clamp(min=1.0)
torsion_loss = torch.sum(torsion_dist_sq * mask, dim=(0, 2, 3)) / torsion_normalizer
# Algorithm 27 line 4: L_anglenorm = mean_{i,f} |||α_tilde_i^f|| - 1|.
# Covers all 7 torsions regardless of mask — this regulariser keeps the
# raw network output close to unit norm before the α̂ = α̃ / ||α̃||
# normalization, independent of whether the torsion is supervised.
angle_norm = torch.sqrt(torch.sum(unnormalized_torsion_angles ** 2, dim=-1) + 1e-8)
if seq_mask is not None:
angle_norm_mask = seq_mask.unsqueeze(0).unsqueeze(-1).expand_as(angle_norm)
else:
angle_norm_mask = torch.ones_like(angle_norm)
norm_normalizer = angle_norm_mask.sum(dim=(0, 2, 3)).clamp(min=1.0)
angle_norm_loss = torch.sum(torch.abs(angle_norm - 1.0) * angle_norm_mask, dim=(0, 2, 3)) / norm_normalizer
return self.torsion_weight * torsion_loss + self.angle_norm_weight * angle_norm_lossThe loss has two terms: between the predicted pair and the ground truth pair, plus a penalty that encourages the predicted pair to lie on the unit circle, meaning . The unit-circle penalty matters because the model’s raw output is a 2D vector that gets normalized before being interpreted as an angle. Keeping the raw output close to unit norm stabilizes the learning dynamics.
12.5 pLDDT, distogram, experimentally-resolved, violations
Four more losses round out the full objective. Most are classification losses attached to the auxiliary heads from §11, plus one physical-plausibility loss.
class PLDDTLoss(torch.nn.Module):
"""Model-confidence loss L_conf (supplement 1.9.6, Algorithm 29 line 4).
Cross-entropy between the predicted pLDDT distribution (from Algorithm 29
lines 1-2) and the one-hot discretisation of the per-residue true lDDT-Cα
score into 50 bins of width 2. The true lDDT-Cα is computed in
`AlphaFoldLoss.compute_loss_terms`, this module just performs the CE.
`filter_by_resolution` zeros the loss on examples whose crystal-structure
resolution falls outside [0.1 Å, 3.0 Å] per supplement 1.9.6.
"""
def __init__(
self,
*,
filter_by_resolution: bool = False,
min_resolution: float = 0.1,
max_resolution: float = 3.0,
):
super().__init__()
self.filter_by_resolution = filter_by_resolution
self.min_resolution = min_resolution
self.max_resolution = max_resolution
def forward(
self,
pred_plddt: torch.Tensor,
true_plddt: torch.Tensor,
seq_mask: Optional[torch.Tensor] = None,
resolution: Optional[torch.Tensor] = None,
):
# pred_plddt, true_plddt: (batch, N_res, n_plddt_bins), latter one-hot.
log_pred = torch.log_softmax(pred_plddt, dim=-1)
# Algorithm 29 line 4: L_conf = mean_i(p_i^{true LDDT T} · log p_i^pLDDT).
# (The published formula omits the minus sign; cross-entropy is a
# negative log-likelihood, hence the sign here.)
conf_loss = -torch.einsum('bic, bic -> bi', true_plddt, log_pred)
if seq_mask is not None:
conf_loss = conf_loss * seq_mask
conf_loss = conf_loss.sum(dim=-1) / seq_mask.sum(dim=-1).clamp(min=1)
else:
conf_loss = torch.mean(conf_loss, dim=-1)
if self.filter_by_resolution and resolution is not None:
resolution = resolution.to(conf_loss.device, dtype=conf_loss.dtype).reshape(-1)
if resolution.numel() == 1 and conf_loss.numel() != 1:
resolution = resolution.expand_as(conf_loss)
else:
resolution = resolution.reshape(conf_loss.shape)
in_range = (
(resolution >= self.min_resolution)
& (resolution <= self.max_resolution)
).to(conf_loss.dtype)
conf_loss = conf_loss * in_range
return conf_losspLDDT is cross-entropy on 50 lDDT- bins. lDDT is computed by scanning through pairs within a distance cutoff and checking how close the predicted distance is to the true distance. It is a local, alignment-free fidelity measure. The pLDDT head is trained to predict which lDDT bin each residue will fall into. This is a classic “learn to predict your own error” setup, and it gives users the confidence scores they eventually see on predicted structures.
class DistogramLoss(torch.nn.Module):
"""Distogram cross-entropy L_dist (supplement 1.9.8 equation 41).
L_dist = -1/N_res^2 Σ_{i,j} Σ_b y_{ij}^b log p_{ij}^b
where p_{ij}^b comes from the distogram head applied to the symmetrised
pair representation and y_{ij}^b is the one-hot encoding of the true
Cβ-Cβ distance (Cα for glycine) into 64 equal-width bins covering 2-22 Å,
with the final bin catching anything more distant. Target construction
lives in `AlphaFoldLoss.compute_loss_terms`.
When `pair_mask` masks padded residues, the denominator is the number of
*valid* pairs rather than `N_res^2` so variable-length batches behave
sensibly; on a full un-padded crop the two agree.
"""
def __init__(self):
super().__init__()
def forward(self, pred_distograms: torch.Tensor, true_distograms: torch.Tensor,
pair_mask: Optional[torch.Tensor] = None):
# input shapes: (batch, N_res, N_res, num_dist_buckets)
log_pred = torch.log_softmax(pred_distograms, dim=-1)
vals = torch.einsum('bijc, bijc -> bij', true_distograms, log_pred)
if pair_mask is not None:
vals = vals * pair_mask
dist_loss = -vals.sum(dim=(1, 2)) / pair_mask.sum(dim=(1, 2)).clamp(min=1)
else:
dist_loss = -torch.mean(vals, dim=(1, 2))
return dist_lossDistogram is cross-entropy on 64 bins of - distance (or pseudo- for glycines). It reads from , not from the predicted structure, which means the pair representation has to encode pairwise distance information before the Structure Module runs. This makes the distogram head a useful auxiliary signal for the pair stack.
class ExperimentallyResolvedLoss(torch.nn.Module):
"""Experimentally-resolved loss L_exp_resolved (supplement 1.9.10 eq 43).
L_exp_resolved = mean_{(i,a)} (
-y_i^a log p_i^{exp resolved,a}
-(1 - y_i^a) log(1 - p_i^{exp resolved,a}))
Binary cross-entropy per (residue, atom37) slot predicting whether that
atom was resolved in the crystal structure. Only used during fine-tuning
(eq 7 fine-tuning row) and only on examples with resolution in
[0.1 Å, 3.0 Å] (1.9.10).
The `atom37_exists` mask restricts the mean to atom slots that are
defined for the residue type — slots that do not exist (e.g. χ atoms on
ALA) would otherwise inject meaningless BCE terms.
"""
def __init__(
self,
*,
filter_by_resolution: bool = False,
min_resolution: float = 0.1,
max_resolution: float = 3.0,
):
super().__init__()
self.filter_by_resolution = filter_by_resolution
self.min_resolution = min_resolution
self.max_resolution = max_resolution
def forward(
self,
exp_resolved_preds: torch.Tensor,
exp_resolved_true: torch.Tensor,
atom37_exists: torch.Tensor,
resolution: Optional[torch.Tensor] = None,
):
xent = torch.nn.functional.binary_cross_entropy_with_logits(
exp_resolved_preds,
exp_resolved_true,
reduction="none",
)
weighted_xent = xent * atom37_exists
normalizer = atom37_exists.sum(dim=(1, 2)).clamp(min=1.0)
loss = weighted_xent.sum(dim=(1, 2)) / normalizer
if self.filter_by_resolution and resolution is not None:
resolution = resolution.to(loss.device, dtype=loss.dtype).reshape(-1)
if resolution.numel() == 1 and loss.numel() != 1:
resolution = resolution.expand_as(loss)
else:
resolution = resolution.reshape(loss.shape)
in_range = (
(resolution >= self.min_resolution)
& (resolution <= self.max_resolution)
).to(loss.dtype)
loss = loss * in_range
return lossExperimentally-resolved is binary cross-entropy per atom. For each of the 14 possible atoms per residue, the model predicts whether that atom was resolved in the experimental structure. Loops, terminal regions, and some side chains are often missing from crystal structures, and the model needs to learn that “not resolved” is not the same as “does not exist.”
class StructuralViolationLoss(torch.nn.Module):
"""Structural-violation loss L_viol (supplement 1.9.11 equations 44-47).
L_viol = L_bondlength + L_bondangle + L_clash (eq 47)
* `L_bondlength` (eq 44): flat-bottom L1 on inter-residue C-N peptide
bond lengths relative to literature values, tolerance 12 σ_lit.
* `L_bondangle` (eq 45): flat-bottom L1 on the cosine of each peptide
bond angle (CA-C-N and C-N-CA) against literature, tolerance 12 σ_lit.
Supplement 1.9.11 describes a single bond-angle term; we sum the two
peptide-bond angles, which is a sum of two paper-faithful flat-bottom
L1 terms and matches what the DeepMind reference releases compute.
* `L_clash` (eq 46): one-sided flat-bottom on VDW overlaps between
non-bonded heavy atoms with tolerance 1.5 Å. Split into between- and
within-residue halves for memory, averaged per atom so the term sits
at a sane scale relative to L_bondlength / L_bondangle (eq 7 applies
an absolute weight of 1.0 to L_viol).
Used only during fine-tuning (eq 7 fine-tuning row).
"""
# Declaring registered buffers as class-level Tensor annotations lets type
# checkers see them as tensors rather than the parent ``Module``.
vdw_table: torch.Tensor
distance_lower_bound_table: torch.Tensor
distance_upper_bound_table: torch.Tensor
def __init__(
self,
violation_tolerance_factor: float = 12.0,
clash_overlap_tolerance: float = 1.5,
):
super().__init__()
bounds = make_atom14_dists_bounds(
overlap_tolerance=clash_overlap_tolerance,
bond_length_tolerance_factor=violation_tolerance_factor,
)
self.register_buffer('vdw_table', torch.tensor(restype_atom14_vdw_radius))
self.register_buffer('distance_lower_bound_table', torch.tensor(bounds["lower_bound"]))
self.register_buffer('distance_upper_bound_table', torch.tensor(bounds["upper_bound"]))
self.violation_tolerance_factor = violation_tolerance_factor
self.clash_overlap_tolerance = clash_overlap_tolerance
def forward(
self,
predicted_positions, # (batch, N_res, 14, 3) — all-atom coordinates
atom_mask, # (batch, N_res, 14) — 1 if atom exists, 0 otherwise
residue_types, # (batch, N_res) — integer residue type index (0–20)
residue_index, # (batch, N_res)
):
connection_violations = self.between_residue_bond_and_angle_loss(
predicted_positions,
atom_mask,
residue_types,
residue_index,
)
between_residue_clashes = self.between_residue_clash_loss(
predicted_positions,
atom_mask,
residue_types,
residue_index,
)
within_residue_violations = self.within_residue_violation_loss(
predicted_positions,
atom_mask,
residue_types,
)
num_atoms = torch.sum(atom_mask, dim=(1, 2)).clamp(min=1e-6)
per_atom_clash = (
between_residue_clashes["per_atom_loss_sum"]
+ within_residue_violations["per_atom_loss_sum"]
)
clash_loss = torch.sum(per_atom_clash, dim=(1, 2)) / num_atoms
return (
connection_violations["c_n_loss_mean"]
+ connection_violations["ca_c_n_loss_mean"]
+ connection_violations["c_n_ca_loss_mean"]
+ clash_loss
)
def between_residue_bond_and_angle_loss(
self,
predicted_positions, # (batch, N_res, 14, 3) — all-atom coordinates
atom_mask, # (batch, N_res, 14) — 1 if atom exists, 0 otherwise
residue_types, # (batch, N_res) — integer residue type index (0–20)
residue_index, # (batch, N_res)
):
eps = 1e-6
this_ca_pos = predicted_positions[:, :-1, 1, :]
this_ca_mask = atom_mask[:, :-1, 1]
this_c_pos = predicted_positions[:, :-1, 2, :]
this_c_mask = atom_mask[:, :-1, 2]
next_n_pos = predicted_positions[:, 1:, 0, :]
next_n_mask = atom_mask[:, 1:, 0]
next_ca_pos = predicted_positions[:, 1:, 1, :]
next_ca_mask = atom_mask[:, 1:, 1]
has_no_gap_mask = (residue_index[:, 1:] - residue_index[:, :-1] == 1).to(predicted_positions.dtype)
c_n_bond_length = torch.sqrt(torch.sum((this_c_pos - next_n_pos) ** 2, dim=-1) + eps)
next_is_proline = (residue_types[:, 1:] == 14).to(predicted_positions.dtype)
gt_length = (
(1.0 - next_is_proline) * between_res_bond_length_c_n[0]
+ next_is_proline * between_res_bond_length_c_n[1]
)
gt_stddev = (
(1.0 - next_is_proline) * between_res_bond_length_stddev_c_n[0]
+ next_is_proline * between_res_bond_length_stddev_c_n[1]
)
c_n_mask = this_c_mask * next_n_mask * has_no_gap_mask
c_n_error = torch.sqrt((c_n_bond_length - gt_length) ** 2 + eps)
c_n_loss_per_residue = torch.relu(c_n_error - self.violation_tolerance_factor * gt_stddev)
c_n_loss = torch.sum(c_n_mask * c_n_loss_per_residue, dim=-1) / (torch.sum(c_n_mask, dim=-1) + eps)
c_n_violation_mask = c_n_mask * (c_n_error > (self.violation_tolerance_factor * gt_stddev))
ca_c_bond_length = torch.sqrt(torch.sum((this_ca_pos - this_c_pos) ** 2, dim=-1) + eps)
n_ca_bond_length = torch.sqrt(torch.sum((next_n_pos - next_ca_pos) ** 2, dim=-1) + eps)
c_ca_unit_vec = (this_ca_pos - this_c_pos) / ca_c_bond_length[..., None]
c_n_unit_vec = (next_n_pos - this_c_pos) / c_n_bond_length[..., None]
n_ca_unit_vec = (next_ca_pos - next_n_pos) / n_ca_bond_length[..., None]
ca_c_n_metric = torch.sum(c_ca_unit_vec * c_n_unit_vec, dim=-1)
ca_c_n_mask = this_ca_mask * this_c_mask * next_n_mask * has_no_gap_mask
ca_c_n_error = torch.sqrt((ca_c_n_metric - between_res_cos_angles_ca_c_n[0]) ** 2 + eps)
ca_c_n_loss_per_residue = torch.relu(
ca_c_n_error - self.violation_tolerance_factor * between_res_cos_angles_ca_c_n[1]
)
ca_c_n_loss = torch.sum(ca_c_n_mask * ca_c_n_loss_per_residue, dim=-1) / (
torch.sum(ca_c_n_mask, dim=-1) + eps
)
ca_c_n_violation_mask = ca_c_n_mask * (
ca_c_n_error > (self.violation_tolerance_factor * between_res_cos_angles_ca_c_n[1])
)
c_n_ca_metric = torch.sum((-c_n_unit_vec) * n_ca_unit_vec, dim=-1)
c_n_ca_mask = this_c_mask * next_n_mask * next_ca_mask * has_no_gap_mask
c_n_ca_error = torch.sqrt((c_n_ca_metric - between_res_cos_angles_c_n_ca[0]) ** 2 + eps)
c_n_ca_loss_per_residue = torch.relu(
c_n_ca_error - self.violation_tolerance_factor * between_res_cos_angles_c_n_ca[1]
)
c_n_ca_loss = torch.sum(c_n_ca_mask * c_n_ca_loss_per_residue, dim=-1) / (
torch.sum(c_n_ca_mask, dim=-1) + eps
)
c_n_ca_violation_mask = c_n_ca_mask * (
c_n_ca_error > (self.violation_tolerance_factor * between_res_cos_angles_c_n_ca[1])
)
per_residue_loss_sum = c_n_loss_per_residue + ca_c_n_loss_per_residue + c_n_ca_loss_per_residue
per_residue_loss_sum = 0.5 * (
torch.nn.functional.pad(per_residue_loss_sum, (0, 1))
+ torch.nn.functional.pad(per_residue_loss_sum, (1, 0))
)
violation_mask = torch.max(
torch.stack(
[c_n_violation_mask, ca_c_n_violation_mask, c_n_ca_violation_mask],
dim=-1,
),
dim=-1,
).values
violation_mask = torch.maximum(
torch.nn.functional.pad(violation_mask, (0, 1)),
torch.nn.functional.pad(violation_mask, (1, 0)),
)
return {
"c_n_loss_mean": c_n_loss,
"ca_c_n_loss_mean": ca_c_n_loss,
"c_n_ca_loss_mean": c_n_ca_loss,
"per_residue_loss_sum": per_residue_loss_sum,
"per_residue_violation_mask": violation_mask,
}
def between_residue_clash_loss(
self,
predicted_positions, # (batch, N_res, 14, 3) — all-atom coordinates
atom_mask, # (batch, N_res, 14) — 1 if atom exists, 0 otherwise
residue_types, # (batch, N_res) — integer residue type index (0–20)
residue_index, # (batch, N_res)
):
batch, N_res = predicted_positions.shape[:2]
overlap_tolerance = self.clash_overlap_tolerance
# Flatten atoms: (batch, N_res*14, 3) and (batch, N_res*14)
pos_flat = predicted_positions.reshape(batch, N_res * 14, 3)
mask_flat = atom_mask.reshape(batch, N_res * 14)
# VDW radii per atom: look up from registered buffer
# residue_types: (batch, N_res) -> vdw: (batch, N_res, 14)
residue_types_clamped = residue_types.clamp(max=20)
vdw = self.vdw_table[residue_types_clamped] # (batch, N_res, 14)
vdw_flat = vdw.reshape(batch, N_res * 14) # (batch, N_res*14)
# Pairwise distances: (batch, N_res*14, N_res*14)
diff = pos_flat[:, :, None, :] - pos_flat[:, None, :, :] # (batch, M, M, 3)
dist = torch.sqrt(torch.sum(diff ** 2, dim=-1) + 1e-8) # (batch, M, M)
# Pair mask: both atoms valid
pair_mask = mask_flat[:, :, None] * mask_flat[:, None, :] # (batch, M, M)
atom_residue_index = residue_index.repeat_interleave(14, dim=1)
unique_residue_pairs = (atom_residue_index[:, :, None] < atom_residue_index[:, None, :]).to(
predicted_positions.dtype
)
pair_mask = pair_mask * unique_residue_pairs
atom_slot_index = torch.arange(14, device=predicted_positions.device).repeat(N_res)
atom_slot_index = atom_slot_index.unsqueeze(0).expand(batch, -1)
residue_type_flat = residue_types.repeat_interleave(14, dim=1)
c_n_bond = (
(atom_slot_index[:, :, None] == 2)
& (atom_slot_index[:, None, :] == 0)
& (atom_residue_index[:, None, :] - atom_residue_index[:, :, None] == 1)
) | (
(atom_slot_index[:, :, None] == 0)
& (atom_slot_index[:, None, :] == 2)
& (atom_residue_index[:, :, None] - atom_residue_index[:, None, :] == 1)
)
disulfide_bond = (
(residue_type_flat[:, :, None] == 4)
& (residue_type_flat[:, None, :] == 4)
& (atom_slot_index[:, :, None] == 5)
& (atom_slot_index[:, None, :] == 5)
)
pair_mask = pair_mask * (~c_n_bond).to(predicted_positions.dtype) * (~disulfide_bond).to(predicted_positions.dtype)
# Overlap: vdw_i + vdw_j - tolerance - dist
vdw_sum = vdw_flat[:, :, None] + vdw_flat[:, None, :] # (batch, M, M)
overlap = vdw_sum - overlap_tolerance - dist
clash = torch.clamp(overlap, min=0) * pair_mask
mean_loss = torch.sum(clash, dim=(1, 2)) / torch.sum(pair_mask, dim=(1, 2)).clamp(min=1e-6)
per_atom_loss_sum = (torch.sum(clash, dim=1) + torch.sum(clash, dim=2)).reshape(batch, N_res, 14)
clash_mask = pair_mask * (dist < (vdw_sum - overlap_tolerance))
per_atom_clash_mask = torch.maximum(
torch.amax(clash_mask, dim=1),
torch.amax(clash_mask, dim=2),
).reshape(batch, N_res, 14)
per_atom_num_clash = (torch.sum(clash_mask, dim=1) + torch.sum(clash_mask, dim=2)).reshape(batch, N_res, 14)
return {
"mean_loss": mean_loss,
"per_atom_loss_sum": per_atom_loss_sum,
"per_atom_clash_mask": per_atom_clash_mask,
"per_atom_num_clash": per_atom_num_clash,
}
def within_residue_violation_loss(
self,
predicted_positions, # (batch, N_res, 14, 3)
atom_mask, # (batch, N_res, 14)
residue_types, # (batch, N_res)
):
residue_types_clamped = residue_types.clamp(max=20)
lower_bound = self.distance_lower_bound_table[residue_types_clamped]
upper_bound = self.distance_upper_bound_table[residue_types_clamped]
distances = torch.sqrt(
torch.sum(
(predicted_positions[:, :, :, None, :] - predicted_positions[:, :, None, :, :]) ** 2,
dim=-1,
) + 1e-8
)
pair_mask = atom_mask[:, :, :, None] * atom_mask[:, :, None, :]
eye = torch.eye(14, device=predicted_positions.device, dtype=predicted_positions.dtype)
pair_mask = pair_mask * (1.0 - eye.view(1, 1, 14, 14))
bound_mask = ((lower_bound > 0.0) | (upper_bound > 0.0)).to(predicted_positions.dtype)
pair_mask = pair_mask * bound_mask
lower_violation = torch.clamp(lower_bound - distances, min=0.0)
upper_violation = torch.clamp(distances - upper_bound, min=0.0)
loss = (lower_violation + upper_violation) * pair_mask
per_atom_loss_sum = torch.sum(loss, dim=-2) + torch.sum(loss, dim=-1)
violations = pair_mask * ((distances < lower_bound) | (distances > upper_bound)).to(predicted_positions.dtype)
per_atom_violations = torch.maximum(
torch.amax(violations, dim=-2),
torch.amax(violations, dim=-1),
)
per_atom_num_clash = torch.sum(violations, dim=-2) + torch.sum(violations, dim=-1)
return {
"per_atom_loss_sum": per_atom_loss_sum,
"per_atom_violations": per_atom_violations,
"per_atom_num_clash": per_atom_num_clash,
}Structural violations is the physicality constraint. It has three components: bond length deviations, bond angle deviations, and van der Waals clashes. Each component is a soft penalty on physically implausible geometry. In the paper training recipe, this loss is used during fine-tuning rather than from the start, because early in training the structures are too wrong for “avoid clashes” to be a productive constraint.
12.6 The combined loss
All of the above land in a single weighted sum:
class AlphaFoldLoss(torch.nn.Module):
"""Combined AlphaFold loss (supplement 1.9 equation 7).
Training:
L = 0.5 L_FAPE + 0.5 L_aux + 0.3 L_dist + 2.0 L_msa + 0.01 L_conf
Fine-tuning (equation 7 fine-tuning row + supplement 1.9.7):
L += 0.01 L_exp_resolved + 1.0 L_viol + 0.1 L_pae
Term-by-term mapping (all weights below are *absolute* eq 7 weights, not
relative fractions):
* `sidechain_weight_frac = 0.5` doubles as the weight of L_FAPE (final
all-atom FAPE, Algorithm 20 line 28) AND the weight on the FAPE half
of L_aux (backbone FAPE averaged over iterations, Algorithm 20 line 17).
We decompose `0.5 L_aux = 0.5 L_aux^{FAPE} + 0.5 L_aux^{torsion}` and
sum `0.5 L_FAPE + 0.5 L_aux^{FAPE}` directly.
* `distogram_weight = 0.3` → 0.3 L_dist (supplement 1.9.8, eq 41).
* `msa_weight = 2.0` → 2.0 L_msa (supplement 1.9.9, eq 42).
* `confidence_weight = 0.01` → 0.01 L_conf (supplement 1.9.6, Alg 29).
* `experimentally_resolved_weight = 0.01` → 0.01 L_exp_resolved (1.9.10).
* `structural_violation_weight = 1.0` → 1.0 L_viol (1.9.11, eq 47).
* `tm_score_weight = 0.1` → 0.1 L_pae (supplement 1.9.7, paragraph
following equation 38: "average categorical cross-entropy loss, with
weight 0.1"). Requires `tm_pred` to be passed; otherwise skipped.
`TorsionAngleLoss` pre-applies the outer 0.5 factor on L_aux^{torsion}
and the 0.02 coefficient on L_anglenorm from Algorithm 27, so
`weighted_torsion_loss = torsion_loss` without an additional weight.
`use_clamped_fape` implements supplement 1.11.5: in 90% of training
mini-batches the backbone FAPE is clamped by 10 Å, and unclamped in the
remaining 10%. Passing a float in [0, 1] mixes the two versions with
the given clamped weight; `None` defaults to fully clamped. The side-
chain FAPE is always clamped regardless, so this knob never reaches it.
"""
def __init__(self, finetune: bool = False, use_clamped_fape: Optional[float] = None):
super().__init__()
self.torsion_angle_loss = TorsionAngleLoss()
self.plddt_loss = PLDDTLoss(
filter_by_resolution=True,
min_resolution=0.1,
max_resolution=3.0,
)
self.distogram_loss = DistogramLoss()
self.msa_loss = MSALoss()
self.experimentally_resolved_loss = ExperimentallyResolvedLoss(
filter_by_resolution=True,
min_resolution=0.1,
max_resolution=3.0,
)
self.structural_violation_loss = StructuralViolationLoss()
self.tm_score_loss = TMScoreLoss(
filter_by_resolution=True,
min_resolution=0.1,
max_resolution=3.0,
)
self.backbone_loss = BackboneTrajectoryLoss()
self.sidechain_fape_loss = AllAtomFAPE()
# Equation 7 weights, all absolute (not relative).
self.sidechain_weight_frac = 0.5
self.distogram_weight = 0.3
self.msa_weight = 2.0
self.confidence_weight = 0.01
self.experimentally_resolved_weight = 0.01
self.structural_violation_weight = 1.0
self.tm_score_weight = 0.1 # supplement 1.9.7 (paragraph after eq 38)
self.finetune = finetune
self.use_clamped_fape = use_clamped_fape
def forward(
self,
structure_model_prediction: dict,
true_rotations: torch.Tensor, # (b, N_res, 3, 3)
true_translations: torch.Tensor, # (b, N_res, 3)
true_atom_positions: torch.Tensor, # (b, N_res, 14, 3)
true_atom_mask: torch.Tensor, # (b, N_res, 14)
true_atom_positions_alt: torch.Tensor,
true_atom_mask_alt: torch.Tensor,
true_atom_is_ambiguous: torch.Tensor,
true_torsion_angles: torch.Tensor, # (b, N_res, 7, 2)
true_torsion_angles_alt: torch.Tensor, # (b, N_res, 7, 2)
true_torsion_mask: torch.Tensor, # (b, N_res, 7)
true_rigid_group_frames_R: torch.Tensor,
true_rigid_group_frames_t: torch.Tensor,
true_rigid_group_frames_R_alt: torch.Tensor,
true_rigid_group_frames_t_alt: torch.Tensor,
true_rigid_group_exists: torch.Tensor,
experimentally_resolved_pred: torch.Tensor,
experimentally_resolved_true: torch.Tensor,
experimentally_resolved_exists: torch.Tensor,
masked_msa_pred: torch.Tensor,
masked_msa_target: torch.Tensor,
masked_msa_mask: torch.Tensor,
plddt_pred: torch.Tensor,
distogram_pred: torch.Tensor,
res_types: torch.Tensor, # (b, N_res) integer 0-20
residue_index: torch.Tensor,
seq_mask: Optional[torch.Tensor] = None, # (b, N_res) 1=valid, 0=padding
return_breakdown: bool = False,
resolution: Optional[torch.Tensor] = None,
tm_pred: Optional[torch.Tensor] = None, # (b, N_res, N_res, n_pae_bins)
):
loss_terms = self.compute_loss_terms(
structure_model_prediction=structure_model_prediction,
true_rotations=true_rotations,
true_translations=true_translations,
true_atom_positions=true_atom_positions,
true_atom_mask=true_atom_mask,
true_atom_positions_alt=true_atom_positions_alt,
true_atom_mask_alt=true_atom_mask_alt,
true_atom_is_ambiguous=true_atom_is_ambiguous,
true_torsion_angles=true_torsion_angles,
true_torsion_angles_alt=true_torsion_angles_alt,
true_torsion_mask=true_torsion_mask,
true_rigid_group_frames_R=true_rigid_group_frames_R,
true_rigid_group_frames_t=true_rigid_group_frames_t,
true_rigid_group_frames_R_alt=true_rigid_group_frames_R_alt,
true_rigid_group_frames_t_alt=true_rigid_group_frames_t_alt,
true_rigid_group_exists=true_rigid_group_exists,
experimentally_resolved_pred=experimentally_resolved_pred,
experimentally_resolved_true=experimentally_resolved_true,
experimentally_resolved_exists=experimentally_resolved_exists,
resolution=resolution,
masked_msa_pred=masked_msa_pred,
masked_msa_target=masked_msa_target,
masked_msa_mask=masked_msa_mask,
plddt_pred=plddt_pred,
distogram_pred=distogram_pred,
res_types=res_types,
residue_index=residue_index,
seq_mask=seq_mask,
tm_pred=tm_pred,
)
if return_breakdown:
return loss_terms["loss"], loss_terms
return loss_terms["loss"]
def compute_loss_terms(
self,
structure_model_prediction: dict,
true_rotations: torch.Tensor,
true_translations: torch.Tensor,
true_atom_positions: torch.Tensor,
true_atom_mask: torch.Tensor,
true_atom_positions_alt: torch.Tensor,
true_atom_mask_alt: torch.Tensor,
true_atom_is_ambiguous: torch.Tensor,
true_torsion_angles: torch.Tensor,
true_torsion_angles_alt: torch.Tensor,
true_torsion_mask: torch.Tensor,
true_rigid_group_frames_R: torch.Tensor,
true_rigid_group_frames_t: torch.Tensor,
true_rigid_group_frames_R_alt: torch.Tensor,
true_rigid_group_frames_t_alt: torch.Tensor,
true_rigid_group_exists: torch.Tensor,
experimentally_resolved_pred: torch.Tensor,
experimentally_resolved_true: torch.Tensor,
experimentally_resolved_exists: torch.Tensor,
masked_msa_pred: torch.Tensor,
masked_msa_target: torch.Tensor,
masked_msa_mask: torch.Tensor,
plddt_pred: torch.Tensor,
distogram_pred: torch.Tensor,
res_types: torch.Tensor,
residue_index: torch.Tensor,
seq_mask: Optional[torch.Tensor] = None,
resolution: Optional[torch.Tensor] = None,
tm_pred: Optional[torch.Tensor] = None,
) -> dict[str, torch.Tensor]:
pred_all_frames_R = structure_model_prediction["all_frames_R"] # (batch, N_res, 8, 3, 3)
pred_all_frames_t = structure_model_prediction["all_frames_t"] # (batch, N_res, 8, 3)
atom_coords = structure_model_prediction["atom14_coords"] # (batch, N_res, 14, 3)
atom_mask = structure_model_prediction["atom14_mask"] # (batch, N_res, 14)
# Canonically rename ambiguous sidechains before any atom-derived supervision.
true_atom_positions, true_atom_mask, alt_naming_is_better = select_best_atom14_ground_truth(
atom_coords,
true_atom_positions,
true_atom_mask,
true_atom_positions_alt,
true_atom_mask_alt,
true_atom_is_ambiguous,
)
renamed_rigid_group_frames_R = torch.where(
alt_naming_is_better[:, :, None, None, None] > 0,
true_rigid_group_frames_R_alt,
true_rigid_group_frames_R,
)
renamed_rigid_group_frames_t = torch.where(
alt_naming_is_better[:, :, None, None] > 0,
true_rigid_group_frames_t_alt,
true_rigid_group_frames_t,
)
backbone_mask = true_atom_mask[:, :, 0] * true_atom_mask[:, :, 1] * true_atom_mask[:, :, 2]
backbone_loss = self.backbone_loss(
structure_model_prediction,
true_rotations,
true_translations,
backbone_mask=backbone_mask,
seq_mask=seq_mask,
use_clamped_fape=self.use_clamped_fape,
)
# Side-chain FAPE is always clamped (supplement 1.11.5); use_clamped_fape
# controls only the backbone FAPE trajectory loss above.
sidechain_loss = self.sidechain_fape_loss(
pred_all_frames_R,
pred_all_frames_t,
atom_coords,
atom_mask,
renamed_rigid_group_frames_R,
renamed_rigid_group_frames_t,
true_atom_positions,
true_atom_mask=true_atom_mask,
seq_mask=seq_mask,
frame_mask=true_rigid_group_exists,
)
torsion_loss = self.torsion_angle_loss(
structure_model_prediction["traj_torsion_angles"],
structure_model_prediction["traj_torsion_angles_unnormalized"],
true_torsion_angles,
true_torsion_angles_alt,
true_torsion_mask,
seq_mask=seq_mask,
)
# --- Derive distogram target (supplement 1.9.8) ---
# Targets are the one-hot encoding of Cβ-Cβ distances (Cα for GLY).
is_gly = (res_types == 7) # (batch, N_res)
cb_idx = torch.where(is_gly, 1, 4) # atom14 slots: CA=1, CB=4
cb_pos = torch.gather(
true_atom_positions, 2,
cb_idx[:, :, None, None].expand(-1, -1, 1, 3),
).squeeze(2)
n_dist_bins = distogram_pred.shape[-1]
cb_mask = torch.gather(true_atom_mask, 2, cb_idx[:, :, None]).squeeze(-1)
distogram_true = distance_bin(cb_pos, n_dist_bins)
dist_pair_mask = cb_mask[:, :, None] * cb_mask[:, None, :]
if seq_mask is not None:
dist_pair_mask = dist_pair_mask * (seq_mask[:, :, None] * seq_mask[:, None, :])
dist_loss = self.distogram_loss(distogram_pred, distogram_true, pair_mask=dist_pair_mask)
msa_loss = self.msa_loss(masked_msa_pred, masked_msa_target, masked_msa_mask)
# --- Derive pLDDT target (supplement 1.9.6) ---
# Compute per-residue lDDT-Cα of the prediction against ground truth,
# then discretise into 50 bins of width 2 (v_bins in Algorithm 29).
# lDDT-Cα is the mean over 4 thresholds (0.5, 1, 2, 4 Å) of the fraction
# of included Cα-Cα distance pairs that are preserved within tolerance.
# "Included" = pairs with d_true < 15 Å, excluding self.
N_res = atom_coords.shape[1]
with torch.no_grad():
pred_ca = atom_coords[:, :, 1, :] # (batch, N_res, 3)
true_ca = true_atom_positions[:, :, 1, :]
true_ca_mask = true_atom_mask[:, :, 1]
true_ca_dists = torch.cdist(true_ca, true_ca) # (batch, N_res, N_res)
pred_ca_dists = torch.cdist(pred_ca, pred_ca)
inclusion = (true_ca_dists < 15.0).float() * (
1.0 - torch.eye(N_res, device=pred_ca.device).unsqueeze(0))
inclusion = inclusion * (true_ca_mask[:, :, None] * true_ca_mask[:, None, :])
if seq_mask is not None:
pair_valid = seq_mask[:, :, None] * seq_mask[:, None, :] # (batch, N_res, N_res)
inclusion = inclusion * pair_valid
dist_error = torch.abs(pred_ca_dists - true_ca_dists)
# Average fraction of preserved distances across four thresholds
lddt = torch.zeros(pred_ca.shape[:2], device=pred_ca.device) # (batch, N_res)
n_included = inclusion.sum(dim=-1).clamp(min=1)
for thresh in [0.5, 1.0, 2.0, 4.0]:
lddt = lddt + ((dist_error < thresh).float() * inclusion).sum(dim=-1) / n_included
lddt = lddt / 4.0 # (batch, N_res) in [0, 1]
lddt_mask = true_ca_mask if seq_mask is None else true_ca_mask * seq_mask
n_plddt_bins = plddt_pred.shape[-1]
plddt_edges = torch.arange(1, n_plddt_bins, device=pred_ca.device).float() / n_plddt_bins
plddt_bin_idx = torch.bucketize(lddt, plddt_edges)
plddt_true = torch.nn.functional.one_hot(plddt_bin_idx, n_plddt_bins).float()
plddt_loss = self.plddt_loss(
plddt_pred,
plddt_true,
seq_mask=lddt_mask,
resolution=resolution,
)
# Equation 7, training row. `backbone_loss` is L_aux^{FAPE} = mean_l(FAPE^l);
# `sidechain_loss` is L_FAPE (all-atom, final layer); `torsion_loss` already
# bundles 0.5 * L_aux^{torsion} + 0.01 * L_aux^{anglenorm} per Algorithm 27
# and the L_aux factor from equation 7.
weighted_backbone_loss = (1.0 - self.sidechain_weight_frac) * backbone_loss
weighted_sidechain_fape_loss = self.sidechain_weight_frac * sidechain_loss
weighted_torsion_loss = torsion_loss
fape_loss = weighted_backbone_loss + weighted_sidechain_fape_loss
structure_loss = fape_loss + weighted_torsion_loss
weighted_distogram_loss = self.distogram_weight * dist_loss
weighted_msa_loss = self.msa_weight * msa_loss
weighted_plddt_loss = self.confidence_weight * plddt_loss
loss = structure_loss + weighted_distogram_loss + weighted_msa_loss + weighted_plddt_loss
loss_terms = {
"loss": loss,
"structure_loss": structure_loss,
"fape_loss": fape_loss,
"backbone_loss": backbone_loss,
"sidechain_fape_loss": sidechain_loss,
"torsion_loss": torsion_loss,
"distogram_loss": dist_loss,
"msa_loss": msa_loss,
"plddt_loss": plddt_loss,
"weighted_backbone_loss": weighted_backbone_loss,
"weighted_sidechain_fape_loss": weighted_sidechain_fape_loss,
"weighted_torsion_loss": weighted_torsion_loss,
"weighted_distogram_loss": weighted_distogram_loss,
"weighted_msa_loss": weighted_msa_loss,
"weighted_plddt_loss": weighted_plddt_loss,
}
if self.finetune:
structural_violation_loss = self.structural_violation_loss(
atom_coords,
atom_mask,
res_types,
residue_index,
)
weighted_structural_violation_loss = self.structural_violation_weight * structural_violation_loss
loss = loss + weighted_structural_violation_loss
loss_terms["structural_violation_loss"] = structural_violation_loss
loss_terms["weighted_structural_violation_loss"] = weighted_structural_violation_loss
exp_resolved_loss = self.experimentally_resolved_loss(
experimentally_resolved_pred,
experimentally_resolved_true,
experimentally_resolved_exists,
resolution=resolution,
)
weighted_exp_resolved_loss = self.experimentally_resolved_weight * exp_resolved_loss
loss = loss + weighted_exp_resolved_loss
loss_terms["experimentally_resolved_loss"] = exp_resolved_loss
loss_terms["weighted_experimentally_resolved_loss"] = weighted_exp_resolved_loss
# Supplement 1.9.7: predicted aligned error / pTM head, fine-tuning
# only, weight 0.1. Skipped silently if tm_pred is not supplied.
if tm_pred is not None:
tm_score_loss = self.tm_score_loss(
tm_pred,
predicted_rotations=structure_model_prediction["final_rotations"],
predicted_translations=structure_model_prediction["final_translations"],
true_rotations=true_rotations,
true_translations=true_translations,
backbone_mask=backbone_mask,
seq_mask=seq_mask,
resolution=resolution,
)
weighted_tm_score_loss = self.tm_score_weight * tm_score_loss
loss = loss + weighted_tm_score_loss
loss_terms["tm_score_loss"] = tm_score_loss
loss_terms["weighted_tm_score_loss"] = weighted_tm_score_loss
loss_terms["loss"] = loss
return loss_termsThe supplement gives specific weights. During the main training stage, the loss is:
During fine-tuning, the experimentally-resolved and structural-violation losses are added with weights and , respectively. The whole thing becomes one scalar loss that gets differentiated end-to-end.
FAPE is the clearest example in the whole AF2 paper of an architectural choice expressed as a loss rather than as a layer. You could imagine building a model that outputs atom coordinates and then applies some canonical alignment (like Kabsch superposition) before comparing to ground truth in a single global frame — and some earlier structure prediction work did exactly this. FAPE skips the alignment step entirely by constructing a loss that is intrinsically alignment-free. Every one of the network’s intermediate frames is supervised by this loss, and because the loss itself is SE(3)-invariant, the frames can live in any global orientation the model finds convenient during training.
This is the same kind of move as, say, choosing a contrastive loss over a classification loss in representation learning — you don’t change the model, you change what “being right” means, and a lot of downstream architectural choices become easier as a consequence. If you take one thing away from the losses section, let it be that FAPE is a load-bearing conceptual move, not a minor implementation detail.
13. Training
The training recipe is where the ambition of AlphaFold2 becomes most obvious. The architecture and loss are one thing; making them converge at scale on messy biological data is another. minAlphaFold2 covers most of the training mechanics we need for a faithful pedagogical implementation — cropping, recycling sampling, feature construction, collation, gradient checkpointing — but it skips one major piece: self-distillation. We will call that out honestly.
13.1 Feature engineering
We saw the input feature builders back in §2: build_target_feat , build_msa_feat , build_extra_msa_feat , and build_template_pair_feat . The training-time additions are the supervision builders, which turn raw .mmcif files into all the targets required by the losses in §12:
def build_processed_example(
example: Dict[str, Any],
*,
crop_size: int,
msa_depth: int,
extra_msa_depth: int,
max_templates: int,
training: bool,
block_delete_training_msa: bool = True,
block_delete_msa_fraction: float = 0.3,
block_delete_msa_randomize_num_blocks: bool = False,
block_delete_msa_num_blocks: int = 5,
masked_msa_probability: float = 0.15,
random_seed: int | None = None,
) -> Dict[str, Any]:
torch_generator = _make_torch_generator(random_seed)
cropped = crop_example(example, crop_size=crop_size, training=training, torch_generator=torch_generator)
return build_processed_example_from_cropped(
cropped,
msa_depth=msa_depth,
extra_msa_depth=extra_msa_depth,
max_templates=max_templates,
training=training,
block_delete_training_msa=block_delete_training_msa,
block_delete_msa_fraction=block_delete_msa_fraction,
block_delete_msa_randomize_num_blocks=block_delete_msa_randomize_num_blocks,
block_delete_msa_num_blocks=block_delete_msa_num_blocks,
masked_msa_probability=masked_msa_probability,
random_seed=random_seed,
)This wrapper crops the raw example and delegates to the cropped-example builder. Downstream of that call, the data path extracts atom14 coordinates and masks from the experimental structure, builds ground-truth backbone frames via rigid_frame_from_three_points, computes torsion angles from ///, and produces the supervised tensors consumed by the losses. The output is a single processed-example dictionary that flows through the rest of the pipeline.
13.2 Cropping
Real proteins can be thousands of residues long. The full Evoformer at paper-spec config, with 48 blocks and triangle multiplication’s scaling, becomes extremely expensive as grows. AlphaFold2’s answer is to train on crops: random contiguous windows of a fixed length , usually 256 residues, sampled from each protein.
def crop_example(
example: Dict[str, Any],
crop_size: int,
training: bool,
*,
torch_generator: torch.Generator | None = None,
) -> Dict[str, Any]:
"""Crop every residue-indexed field of ``example`` to ``crop_size``.
Supplement 1.2.8: during training all per-example fields with a
residue axis are cropped to a single contiguous region of ``N_res =
crop_size`` residues. The start is picked by ``_crop_start``. Chains
shorter than ``crop_size`` pass through unchanged.
"""
length = int(example["aatype"].shape[0])
assert example["msa"].shape[1] == length, (
f"msa residue axis {example['msa'].shape[1]} must match aatype length {length}"
)
if length <= crop_size:
cropped = dict(example)
cropped["crop_start"] = 0
return cropped
start = _crop_start(length, crop_size=crop_size, training=training, torch_generator=torch_generator)
end = start + crop_size
residue_slice = slice(start, end)
cropped = dict(example)
cropped["crop_start"] = start
# Residue axis only: (N_res,) -> (crop_size,)
cropped["aatype"] = example["aatype"][residue_slice]
# MSA residue axis: (N_seq, N_res) -> (N_seq, crop_size)
cropped["msa"] = example["msa"][:, residue_slice]
cropped["deletions"] = example["deletions"][:, residue_slice]
if "between_segment_residues" in example:
cropped["between_segment_residues"] = example["between_segment_residues"][residue_slice]
if "residue_index" in example:
cropped["residue_index"] = example["residue_index"][residue_slice]
# Template residue axis: (N_templ, N_res, ...) -> (N_templ, crop_size, ...)
cropped["template_aatype"] = example["template_aatype"][:, residue_slice]
cropped["template_atom14_positions"] = example["template_atom14_positions"][:, residue_slice]
cropped["template_atom14_mask"] = example["template_atom14_mask"][:, residue_slice]
# Supervision residue axis: (N_res, atom14, ...) -> (crop_size, atom14, ...)
cropped["atom14_positions"] = example["atom14_positions"][residue_slice]
cropped["atom14_mask"] = example["atom14_mask"][residue_slice]
return croppedThe mechanics are simple: if the chain is shorter than the crop size, pass it through unchanged. Otherwise, pick one contiguous residue window and slice every residue-indexed field consistently: target sequence, MSA columns, template coordinates, atom14 supervision, and masks. During training, the start index is sampled uniformly by _crop_start; at inference, it is centered deterministically.
This has a nontrivial implication. The model often trains on pieces of proteins but must generalize to full-length proteins at inference. Empirically, the triangle-consistency inductive biases in the Evoformer and the geometry of the Structure Module are strong enough that local structural relationships learned on crops compose reasonably well.
13.3 Batch collation
With variable-length crops, since some proteins are shorter than the crop size, we need to pad and mask. collate_batch does both:
def collate_batch(
examples: List[Dict[str, Any]],
*,
crop_size: int,
msa_depth: int,
extra_msa_depth: int,
max_templates: int,
training: bool,
block_delete_training_msa: bool = True,
block_delete_msa_fraction: float = 0.3,
block_delete_msa_randomize_num_blocks: bool = False,
block_delete_msa_num_blocks: int = 5,
masked_msa_probability: float = 0.15,
random_seed: int | None = None,
num_recycling_samples: int = 1,
num_ensemble_samples: int = 1,
) -> Dict[str, Any]:
"""Collate a list of raw examples into a padded batch dict.
Pipeline per example: crop → build features/labels → pad to batch max.
All residue-indexed fields are padded with zeros to ``max_length``
(longest crop in the batch), MSA-indexed fields to ``max_cluster`` /
``max_extra``, and template-indexed fields to ``max_templates_in_batch``.
Padding masks on ``seq_mask`` / ``msa_mask`` / ``extra_msa_mask`` (set by
``build_processed_example_from_cropped`` before padding) propagate
through to the model so the attention and loss layers ignore the padded
slots.
When ``num_recycling_samples > 1`` or ``num_ensemble_samples > 1`` we
additionally pre-sample the MSA-derived features ``num_recycling_samples
× num_ensemble_samples`` times per example and stack them along two
new leading axes of the MSA fields (Algorithm 2 line 4). The model's
``_sampled_feature_slice`` then indexes back into these axes. Deterministic
per-example seeding via ``_example_seed`` lets tests assert identical
batches across runs.
"""
cropped_examples = [
crop_example(
example,
crop_size=crop_size,
training=training,
torch_generator=_make_torch_generator(None if random_seed is None else _example_seed(random_seed, index)),
)
for index, example in enumerate(examples)
]
processed = [
build_processed_example_from_cropped(
cropped,
msa_depth=msa_depth,
extra_msa_depth=extra_msa_depth,
max_templates=max_templates,
training=training,
block_delete_training_msa=block_delete_training_msa,
block_delete_msa_fraction=block_delete_msa_fraction,
block_delete_msa_randomize_num_blocks=block_delete_msa_randomize_num_blocks,
block_delete_msa_num_blocks=block_delete_msa_num_blocks,
masked_msa_probability=masked_msa_probability,
random_seed=None if random_seed is None else _example_seed(random_seed, index),
)
for index, cropped in enumerate(cropped_examples)
]
sampled_msa_features = _build_sampled_msa_features(
cropped_examples,
num_recycling_samples=num_recycling_samples,
num_ensemble_samples=num_ensemble_samples,
msa_depth=msa_depth,
extra_msa_depth=extra_msa_depth,
training=training,
block_delete_training_msa=block_delete_training_msa,
block_delete_msa_fraction=block_delete_msa_fraction,
block_delete_msa_randomize_num_blocks=block_delete_msa_randomize_num_blocks,
block_delete_msa_num_blocks=block_delete_msa_num_blocks,
masked_msa_probability=masked_msa_probability,
random_seed=random_seed,
)
max_length = max(item["aatype"].shape[0] for item in processed)
max_cluster = max(item["msa_feat"].shape[0] for item in processed)
max_extra = max(item["extra_msa_feat"].shape[0] for item in processed)
if sampled_msa_features:
max_cluster = max(
max_cluster,
max(
sample["msa_feat"].shape[0]
for recycle_samples in sampled_msa_features
for ensemble_samples in recycle_samples
for sample in ensemble_samples
),
)
max_extra = max(
max_extra,
max(
sample["extra_msa_feat"].shape[0]
for recycle_samples in sampled_msa_features
for ensemble_samples in recycle_samples
for sample in ensemble_samples
),
)
max_templates_in_batch = max(item["template_pair_feat"].shape[0] for item in processed)
batch: Dict[str, Any] = {"chain_id": [item["chain_id"] for item in processed]}
def stack(key: str, *, fill_value: float = 0.0, target_shape: Sequence[int]) -> None:
batch[key] = torch.stack(
[pad_tensor(item[key], target_shape=target_shape, value=fill_value) for item in processed],
dim=0,
)
padding_shapes = {
# Model inputs.
"aatype": (max_length,),
"resolution": (),
"target_feat": (max_length, TARGET_FEAT_DIM),
"residue_index": (max_length,),
"seq_mask": (max_length,),
# MSA inputs and masked-MSA supervision.
"msa_feat": (max_cluster, max_length, MSA_FEAT_DIM),
"msa_mask": (max_cluster, max_length),
"extra_msa_feat": (max_extra, max_length, EXTRA_MSA_FEAT_DIM),
"extra_msa_mask": (max_extra, max_length),
"masked_msa_target": (max_cluster, max_length, MSA_ALPHABET_SIZE),
"masked_msa_mask": (max_cluster, max_length),
# Template inputs.
"template_pair_feat": (max_templates_in_batch, max_length, max_length, TEMPLATE_PAIR_DIM),
"template_angle_feat": (max_templates_in_batch, max_length, TEMPLATE_ANGLE_DIM),
"template_mask": (max_templates_in_batch,),
"template_residue_mask": (max_templates_in_batch, max_length),
# Structure and auxiliary-head supervision.
"true_rotations": (max_length, 3, 3),
"true_translations": (max_length, 3),
"true_atom_positions": (max_length, 14, 3),
"true_atom_mask": (max_length, 14),
"true_atom_positions_alt": (max_length, 14, 3),
"true_atom_mask_alt": (max_length, 14),
"true_atom_is_ambiguous": (max_length, 14),
"true_torsion_angles": (max_length, 7, 2),
"true_torsion_angles_alt": (max_length, 7, 2),
"true_torsion_mask": (max_length, 7),
"true_rigid_group_frames_R": (max_length, 8, 3, 3),
"true_rigid_group_frames_t": (max_length, 8, 3),
"true_rigid_group_frames_R_alt": (max_length, 8, 3, 3),
"true_rigid_group_frames_t_alt": (max_length, 8, 3),
"true_rigid_group_exists": (max_length, 8),
"atom37_exists": (max_length, atom_type_num),
"experimentally_resolved_true": (max_length, atom_type_num),
"res_types": (max_length,),
"backbone_mask": (max_length,),
"pseudo_beta_mask": (max_length,),
"pseudo_beta_positions": (max_length, 3),
}
for key, target_shape in padding_shapes.items():
stack(key, target_shape=target_shape)
if sampled_msa_features:
def stack_sampled(key: str, *, fill_value: float = 0.0, target_shape: Sequence[int]) -> None:
recycle_batches = []
for recycle_samples in sampled_msa_features:
ensemble_batches = []
for ensemble_samples in recycle_samples:
ensemble_batches.append(
torch.stack(
[
pad_tensor(sample[key], target_shape=target_shape, value=fill_value)
for sample in ensemble_samples
],
dim=0,
)
)
recycle_batches.append(torch.stack(ensemble_batches, dim=0))
batch[key] = torch.stack(recycle_batches, dim=0)
sampled_padding_shapes = {
"msa_feat": (max_cluster, max_length, MSA_FEAT_DIM),
"msa_mask": (max_cluster, max_length),
"extra_msa_feat": (max_extra, max_length, EXTRA_MSA_FEAT_DIM),
"extra_msa_mask": (max_extra, max_length),
"masked_msa_target": (max_cluster, max_length, MSA_ALPHABET_SIZE),
"masked_msa_mask": (max_cluster, max_length),
}
for key in MSA_SAMPLE_FEATURE_KEYS:
stack_sampled(key, target_shape=sampled_padding_shapes[key])
return batchEvery feature tensor in the batch is padded to the largest example in the batch, and a seq_mask tensor is threaded through the attention operations to block out padded positions. Attention scores at padded indices go to before softmax, and outputs at padded positions are ignored before loss computation. This is why masks appear everywhere in minAlphaFold2.
13.4 Recycling sampling
We covered the mechanics of recycling in §9: feed previous-cycle outputs back into current-cycle inputs, then detach gradients between cycles. What changes at training time is that the number of cycles is sampled uniformly:
n_cycles = random.randint(1, max_cycles) # uniform during trainingThis forces the model to produce usable representations at many cycle counts, not just the maximum. A model trained only with 4 cycles could specialize to the 4-cycle case. With uniform cycle sampling, the weights must handle 1-cycle, 2-cycle, …, -cycle inputs reasonably well.
13.5 Gradient checkpointing
The full Evoformer at 48 blocks, with recycling and triangle multiplication’s cost, has an activation memory footprint that can easily dominate the parameter memory. PyTorch’s torch.utils.checkpoint trades compute for memory by not storing intermediate activations during the forward pass. During the backward pass, the forward is re-run for each checkpointed block to reconstruct those activations.
In minAlphaFold2, gradient checkpointing wraps every Evoformer block and every extra-MSA block. The practical effect is simple: spend more compute so you can store fewer activations. For a model where the expensive parts are repeated dozens of times, this is the difference between a runnable training script and one that runs out of memory immediately.
13.6 The honest gap: self-distillation
The full AlphaFold2 training procedure has one piece that minAlphaFold2 does not implement: self-distillation. In the full procedure, an undistilled model is trained on known PDB structures. That model is then used to predict structures for large numbers of unlabeled protein sequences from Uniclust30. High-confidence predicted structures are retained as pseudo-ground-truth and mixed with real PDB structures during training.
One important detail: in the supplement, the filtering metric for the distillation set is not pLDDT but a KL-based confidence metric computed from predicted aligned error-like distributions; the authors note that pLDDT would likely work as well or better. Either way, the conceptual role is the same. Self-distillation lets the model learn from unlabeled sequence space, but it requires substantial infrastructure: two-stage training, a massive prediction run, and careful filtering of pseudo-labels.
minAlphaFold2 leaves self-distillation as a deliberate gap. Self-distillation matters for final accuracy, but it is mostly a data-generation and training-infrastructure story rather than a new architectural idea. Including it faithfully would add a large amount of pipeline plumbing without changing the conceptual account of Evoformer, recycling, IPA, or FAPE. I am noting the gap here so that nobody confuses this implementation with a fully faithful reproduction of DeepMind’s entire training recipe.
13.7 The training loop itself
Everything above lands in a single training loop:
def train_step(
model: AlphaFold2,
loss_fn: AlphaFoldLoss,
optimizer: torch.optim.Optimizer,
batch: dict[str, Any],
training_config: TrainingConfig,
) -> dict[str, float]:
"""Single forward/backward/step iteration.
Gradient clipping follows supplement 1.11.3 (clip by global norm = 0.1
by default). The paper clips per-example within a mini-batch; we clip
over the whole mini-batch, which is equivalent at ``batch_size = 1``
(the default pedagogical setting).
"""
device = resolve_device(training_config.device)
model.train()
optimizer.zero_grad(set_to_none=True)
batch = move_to_device(batch, device)
outputs = model(**model_inputs_from_batch(batch, training_config))
per_example_loss = loss_fn(**loss_inputs_from_batch(batch, outputs))
loss = per_example_loss.mean()
loss.backward()
if training_config.grad_clip_norm is not None:
torch.nn.utils.clip_grad_norm_(model.parameters(), training_config.grad_clip_norm)
optimizer.step()
return {"loss": float(loss.item())}The loop itself is standard PyTorch: a forward pass through the model with the right n_cycles, an AlphaFoldLoss call on the outputs, .backward(), gradient clipping, and an optimizer step. The learning-rate schedule is warmup plus decay. Most of the interesting work is in the model and loss; the training loop itself fits on one screen.
With that, we have the full pipeline: input features, Evoformer, Structure Module, auxiliary heads, losses, and training procedure. The last two sections make this concrete. §14 walks through a real single-PDB overfit run with a structure viewer, and §15 points at what AlphaFold2 left open.
14. A real-world walkthrough
We have now built up enough machinery. Thirteen sections of architecture, geometry, loss functions, and training details. So what does it look like when you actually run this thing?
The cleanest test, and the one that convinces you the pipeline works end-to-end, is a single-protein overfit. Pick one PDB, feed it to the model as both input sequence and target coordinates, and train until the model memorizes the structure. If the whole pipeline is coherent — features, Evoformer, Structure Module, loss, optimizer — the predicted coordinates should move toward the ground truth even on CPU. If something important is wrong, such as a sign error in a frame update or a loss computed in the wrong coordinate system, the overfit will usually fail. It is a small test, but it exercises almost everything.
14.1 The setup
The protein I’m using is 1CRN, crambin, a 46-residue plant protein with three disulfide bonds. It is small enough to train on a laptop CPU in under a minute, but geometrically nontrivial enough that a random-initialized model gets it very wrong at step 0 and recognizably right by step 600.
The driver script lives at scripts/overfit_single_pdb.py in the minAlphaFold repo. Simplified to the essentials:
parsed = parse_pdb("1crn.pdb")
example = build_minimal_example("1crn", parsed)
batch = collate_batch([example], crop_size=n_res, msa_depth=1,
extra_msa_depth=0, max_templates=0, ...)
model = AlphaFold2(load_model_config("tiny")) # 0.09M params, 1 Evoformer block
loss_fn = AlphaFoldLoss(finetune=False)
optimizer = build_optimizer(model, TrainingConfig(learning_rate=1e-3, ...))
for step in range(1, 601):
optimizer.zero_grad()
outputs = model(**model_inputs_from_batch(batch, tc))
loss = loss_fn(**loss_inputs_from_batch(batch, outputs)).mean()
loss.backward()
torch.nn.utils.clip_grad_norm_(model.parameters(), 0.1)
optimizer.step()A few things are worth flagging about this setup. The MSA has exactly one sequence: the query itself. Templates are disabled. Extra MSA is disabled. We are running the tiny profile, which has about 90k parameters, 1 Evoformer block, and a shallow Structure Module, not full paper-spec AlphaFold2. And we are training with the same sequence and the same target at every step, so there is no generalization happening here. The model is memorizing one structure.
This is what makes the run a pipeline test rather than a science test. If a 90k-parameter model running on one protein for 600 steps converges toward the right geometry, that does not mean AlphaFold2 generalizes. It means the implementation is coherent enough that gradient descent on FAPE can drive coordinates toward ground truth. That is a low bar, but it is a necessary one, and it catches an enormous number of bugs. “Does my loss go down?” is too weak a question. “Does the predicted structure start looking like the real one?” is the real test.
14.2 What actually happens
The widget below loads 11 checkpoints from a real run: the same code as above, executed on CPU, producing a trajectory from random initialization at step 0 to the step-600 prediction. Drag the scrubber, hit play, or click Reset. The gray cartoon is the ground truth. The colored cartoon (or solid red, depending on the toggle) is the prediction at the selected step.
The story the scrubber tells is roughly the following:
- Step 0-10: the prediction is essentially random. Cα RMSD is about 9.7 Å. The Structure Module has produced 46 backbone frames from random weights, so there is no reason yet to expect a protein-like structure.
- Step 10-100: the model starts to contract and organize. FAPE’s gradient is pulling each residue toward consistency with many local-frame views of the ground truth. The prediction is still quite wrong, but it is less diffuse.
- Step 100-300: the structure starts to become recognizable. Around step 145, the prediction begins to look protein-like, and by step 295, Cα RMSD is about 7.4 Å.
- Step 300-600: the model learns the specific fold of 1CRN: a compact mixed alpha/beta structure constrained by three disulfide bonds. By step 600, Cα RMSD has dropped to about 2.25 Å, close enough that the prediction and ground truth visibly overlay.
Total wall time: roughly 30 seconds on a single CPU core. Total parameters: about 90,000, roughly three orders of magnitude smaller than paper-spec AF2. This is what the tiny profile is good for: a smoke test that exercises the model with minimal time and memory.
14.3 What this does and doesn’t tell us
The overfit says: the pipeline is coherent. Features get built correctly, the Evoformer maps them through, the Structure Module produces frames, FAPE computes a useful gradient, the backbone update and all-atom reconstruction compose into plausible coordinates, and optimization drives all of this toward the ground truth. Every major code path from §§2-13 is exercised in a single forward-backward loop, and the final structure looks like a real protein.
It does not say: AlphaFold2 generalizes. The model has seen exactly one protein. Step 600’s 2.25 Å Cα RMSD is memorization, not prediction. Real AlphaFold2 training uses known PDB structures, deep MSAs, templates, self-distillation on large unlabeled sequence sets, and enormous compute. That is a world apart from a laptop CPU and one PDB.
Even so, the overfit is still the most satisfying “does my code actually work?” demo I know of, because it makes progress visible in the domain that matters. A loss curve going down could mean many things. A structure moving from random geometry into crambin means the loss is going down for the right reason.
If you’d like to run it yourself, the command is:
# From the minAlphaFold2 repo root:
python scripts/overfit_single_pdb.py --pdb path/to/1crn.pdb --steps 600 --model-profile tinyIt will write its own PDBs and metrics to artifacts/overfit_single_pdb/1crn/. Watch the per-step log. Seeing Cα RMSD drop in real time does the same job as the widget above, just without the 3D rendering.
15. What AlphaFold2 left unsolved
AlphaFold2 changed structural biology, and like most field-changing results, it also clarified the next set of problems. The paper largely solved monomeric protein structure prediction for proteins with reasonable MSA coverage. That turned out to be an enormous class of proteins. But real biology cares about many things that are not fully captured by “predict the most likely monomer structure from a sequence”:
Protein complexes. Proteins rarely act alone. They form complexes with other proteins, DNA, RNA, small molecules, ions, and membranes. AF2 was built primarily around monomeric protein structure prediction. AlphaFold-Multimer extended the system to complexes, and AlphaFold3 went further with a redesigned architecture, diffusion-based output, and a scope that includes nucleic acids, ligands, and post-translational modifications.
Dynamics. Proteins are not static. They breathe, flex, and transition between conformations that may be functionally distinct. AF2 predicts a single likely structure, which is often enough for structural biology but not always enough for enzyme mechanisms, allosteric regulation, or motion-dependent function. MSA-clustering methods can coax multiple states out of AF2 by perturbing the input MSA, while diffusion-based successors such as AlphaFold3 and Boltz-1 are better suited to sampling.
Design. Structure prediction is the forward problem: sequence → structure. The inverse problem, “give me a sequence that folds into this structure,” is protein design. RoseTTAFold Diffusion, ProteinMPNN, and AlphaProteo all live in or adjacent to this inverse space. They often use AF2-descended ideas to evaluate designs, but the generative problem is different.
Language-model shortcuts. Perhaps the most interesting follow-up is the shift toward models that replace the MSA entirely. ESMFold uses a pretrained protein language model in place of the MSA pipeline, with the implicit claim that a large enough protein transformer has already learned many of the co-evolutionary patterns the MSA was meant to surface. ESM3 goes further by training jointly on sequence, structure, and function, positioning itself as a more general biological foundation model rather than a structure-prediction specialist. I’ve written a multi-part primer on protein language models that traces this arc from AF2 through ESM3 in more detail.
Compute and inference cost. AF2’s training run was enormous, and its per-sequence inference cost is not trivial either, especially when MSA search is included. A lot of real-world work in drug discovery, enzyme engineering, and synthetic biology benefits from cheaper, faster predictions. ESMFold, distillation-based approaches, and newer compute-optimized architectures are all responses to this constraint.
One of the more interesting meta-observations about this space is how many of AF2’s architectural choices turned out to be load-bearing and how many turned out to be replaceable. The triangle updates, IPA-style geometric reasoning, FAPE-like losses, and recycling all proved extremely influential. The specific MSA pipeline is already less central than it looked in 2021. The Structure Module has been rethought in diffusion-based successors. The training recipe has also been reworked.
This is useful to keep in mind when reading any landmark paper. Some components become permanent conceptual machinery for the field. Others are historically important because they made the first working system possible, but later get swapped out. AlphaFold2 is especially worth reading because it contains both kinds of idea.
— end —
If you’ve made it this far, thank you for reading! If you found this useful, I write regularly about AI, biology, and adjacent topics at chrishayduk.com. The annotation project will continue from here. ESM-2, ESMFold, ESM-3, and AlphaFold3 are all coming up soon!